Article Text

Abstract
Introduction A new prognostic model has been developed and externally validated, the aim of which is to assist in the management of the blunt chest wall trauma patient in the emergency department (ED). A definitive randomised controlled trial (impact trial) is required to assess the clinical and cost effectiveness of the new model before it can be accepted in clinical practice. The purpose of this trial is to assess the feasibility and acceptability of such a definitive trial and inform its design.
Methods/analysis This feasibility trial is designed to test the methods of a multicentre, cluster-randomised (stepped- wedge) trial, with a substantial qualitative component. Four EDs in England and Wales will collect data for all blunt chest wall trauma patients over a 5-month period; in the initial period acting as the controls (normal care), and in the second period acting as the interventions (in which the new model will be used). Baseline measurements including completion of the SF-12v2 will be obtained on initial assessment in the ED. Patient outcome data will then be collected for any subsequent hospitalisations. Data collection will conclude with a 6-week follow-up completion of two surveys (SF-12v2 and Client Services Receipt Inventory). Analysis of outcomes will focus on feasibility, acceptability and trial processes and will include recruitment and retention rates, attendance at clinician training rates and use of model in the ED. Qualitative feedback will be obtained through clinician interviews and a research nurse focus group. An evaluation of the feasibility of health economics outcomes data will be completed.
Ethics and dissemination Wales Research Ethics Committee 6 granted approval for the trial in September 2016. Patient recruitment will commence in February 2017. Planned dissemination is through publication in a peer-reviewed Emergency Medicine Journal, presentation at appropriate conferences and to stakeholders at professional meetings.
Trial registration number ISRCTN95571506; Pre-results.
- blunt chest wall trauma
- prognostic model
- feasibility trial
- stepped-wedge randomised trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
The main strength of the trial design is that it will test all of the methodological components of the future definitive trial, including the trial infrastructure, randomisation process, data management system, both qualitative and quantitative analysis, follow-up procedures and a health economic analysis.
The health economic analysis will ensure accurate future funding applications for the full definitive trial.
The qualitative analysis will inform the researchers’ understanding of the use of the risk score by clinicians in daily clinical practice.
The cluster-randomised trial design that needed to be tested in this feasibility trial is limited by the small number of clusters (emergency departments participating). In the future definitive trial, more clusters will be possible due to the greater number of participating sites.
Introduction
Blunt chest wall trauma accounts for >15% of all trauma admissions to emergency departments (ED) worldwide, with reported mortality ranging between 4% and 60%.1–3 The difficulties in the management of the blunt chest wall trauma patient in the ED are becoming increasingly well recognised in the literature.4 5 The blunt chest wall trauma patient commonly presents to the ED with no respiratory difficulties, but can develop complications up to approximately 72 hours after initial presentation.1–4 Clinical symptoms are not considered by most clinicians to be an accurate predictor of outcome following non-life-threatening blunt chest wall trauma.1–7 Decisions regarding the appropriate level of care required by the patient following discharge from the ED are therefore difficult, which is further compounded by the lack of current national guidelines. An improvement in the ability to identify the high-risk blunt chest wall trauma patient on initial assessment in the ED would potentially lead to a reduction in these poor outcomes.8 9
A new prognostic model to assist in the management of blunt chest wall trauma patients in the ED has been developed and externally validated in a large multicentre study.1 5–7 The model comprises five risk factors: age, number of rib fractures, pre-existing chronic lung disease, use of preinjury anticoagulants and oxygen saturation on initial assessment in the ED. The patient is scored on each risk factor and the total score is used to guide the clinician in the ED as to whether the patient should be admitted to the ward, critical care unit or can be safely discharged home (see online supplementary material 1). Guidelines recommend however that prognostic models should not be used in clinical practice until an impact study has been completed, in which the clinical application of the model has been tested.10 11 Prior to undertaking a large definitive randomised controlled trial of the clinical and cost effectiveness of the prognostic model, a smaller-scale trial is required to address the issues of feasibility and acceptability.
Trial aims
The aim of this trial (protocol version 5, 6 January 2017) is to establish the feasibility and acceptability of the final definitive impact trial, which will ultimately determine whether the prognostic model can be used safely and effectively in clinical practice in the UK. The trial objectives are to (1) evaluate the infrastructure necessary to perform a future definitive trial, including the Trial Management Group, Trial Steering/Data Management Committee; (2) test the feasibility of the proposed stepped-wedge, cluster-randomised design; (3) evaluate and qualitatively explore the compliance of the clinicians in using the prognostic model (and reasons for non-compliance); (4) assess the appropriateness of the training manual and consistency of training provided by each principal investigator (PI); (5) quantify the number of patients required for a full definitive trial through the estimation of the magnitude of effect and necessary parameters, including the margin of error acceptable to achieve the proposed outcomes; (6) assess the processes of patient recruitment, consent and reasons for non-participation; (7) assess the quantity and potential patterns of missing data; (8) test the feasibility of collecting the proposed outcome measures for a full trial, including optimal time points, using the electronic case report form on the REDCap data collection system; and (9) decide whether a fully powered, multicentre randomised trial is indicated by formal assessment of feasibility trial findings against pre-set progression criteria.
Methods and analysis
Trial design and randomisation
This feasibility trial is designed to test the methods of a multicentre, prospective, cluster-randomised (stepped-wedge) trial,12 with a substantial qualitative component. Figure 1 outlines the trial procedure. There will be four hospitals participating in the trial, which will be run using a stepped-wedge design. In accordance with Medical Research Council guidance,13 the unit randomisation will be the EDs, rather than patients, since the intervention will be used by ED clinicians, with the aim of studying effects on patient outcomes. All participating EDs will begin as control sites, at the same time, testing conventional management without the model, for a period of 1 month (control arm). Every month, following the initial month’s data collection period as controls, one hospital will be randomly assigned to become an intervention arm (in which all clinicians in the ED will be using the risk model) sequentially, until all hospitals are acting as interventions.

Schematic representation of the trial’s timelines. PI, principal investigator.
The randomisation process applies to the hospitals (known as clusters) participating in the trial, rather than at a patient level. The order in which hospitals EDs convert to intervention status is based on a purely random process (involving only computer-generated random numbers, but no information on the individual hospitals or EDs) and can be regarded as equivalent to the drawing of names at random. As there are four hospitals participating in the trial, all patient recruitment will be completed over a 5-month period. This trial design will test the feasibility of the classic stepped-wedge design to be used in the future definitive trial.
During the control period, the doctors in each ED will receive training on the model from the PI in each ED. Each PI will be instructed at the start of the trial on how to train the clinicians and a training manual will be designed and provided.
Population
Patients will be included in the trial if they present to the ED with isolated blunt chest wall trauma, are aged ≥18 and are capable of giving consent to participation. Potential patients will be excluded if they are under the age of 18, lack capacity to give informed consent, present with any immediately life-threatening injuries or any concurrent injury that will determine the patient’s management (rather than the chest trauma). Patients will be withdrawn from the trial if they lose capacity (including death) to complete the surveys and if they request to be withdrawn. These data will be recorded as part of the assessment of the trial’s success criteria in order to inform the design of the future definitive trial. A list of all patients who decline to participate will also be completed in order that the trial team can assess the recruitment rates at the end of the study.
Setting and recruitment
This multicentre feasibility trial will be run in the EDs of the Royal Gwent Hospital in Newport, Musgrove Park Hospital in Taunton, Salford Royal Hospital and Manchester Royal Infirmary (all large teaching hospitals located in the UK). The clinicians or research nurses in the ED will screen, recruit and consent eligible patients to the trial.
Sample size
A 5-month recruitment period has been proposed. In 2015, >1200 blunt chest wall trauma patients presented to the ED of the hospital of the trial research team, with >100 of these admitted. If the four participating hospitals recruit for 5 months each, between 30 and 80 patients per site should be recruited, allowing for loss to follow-up, low response rates with follow-up surveys and difficulties with recruiting at weekends if research nurses only work Monday to Friday. This sample size is the minimum number of participants considered necessary to test the processes of data collection, based on existing recommendations with respect to the number of patients required to yield meaningful estimates of parameters of interest.14
The analysis will lead to an estimation of the sample size for the definitive trial needed to yield 80% power when using a significance level of 5%, by establishing non-inferiority between use of the prognostic tool and standard care, as revealed by the feasibility study.
The analysis will include descriptive data (means and SD) on all outcomes collected, including levels of missing data, leading to the calculation of clinically important differences. The conventional sample size calculations for the future definitive trial will be developed using this analysis and the consensus among the Trial Management Group and Trial Steering Committee of the minimal clinically important difference in these measures. If the recruitment and retention plan is shown to be optimistic in this feasibility trial, then this is an important finding which will inform the sample size calculation for the future main definitive trial.
Interventions
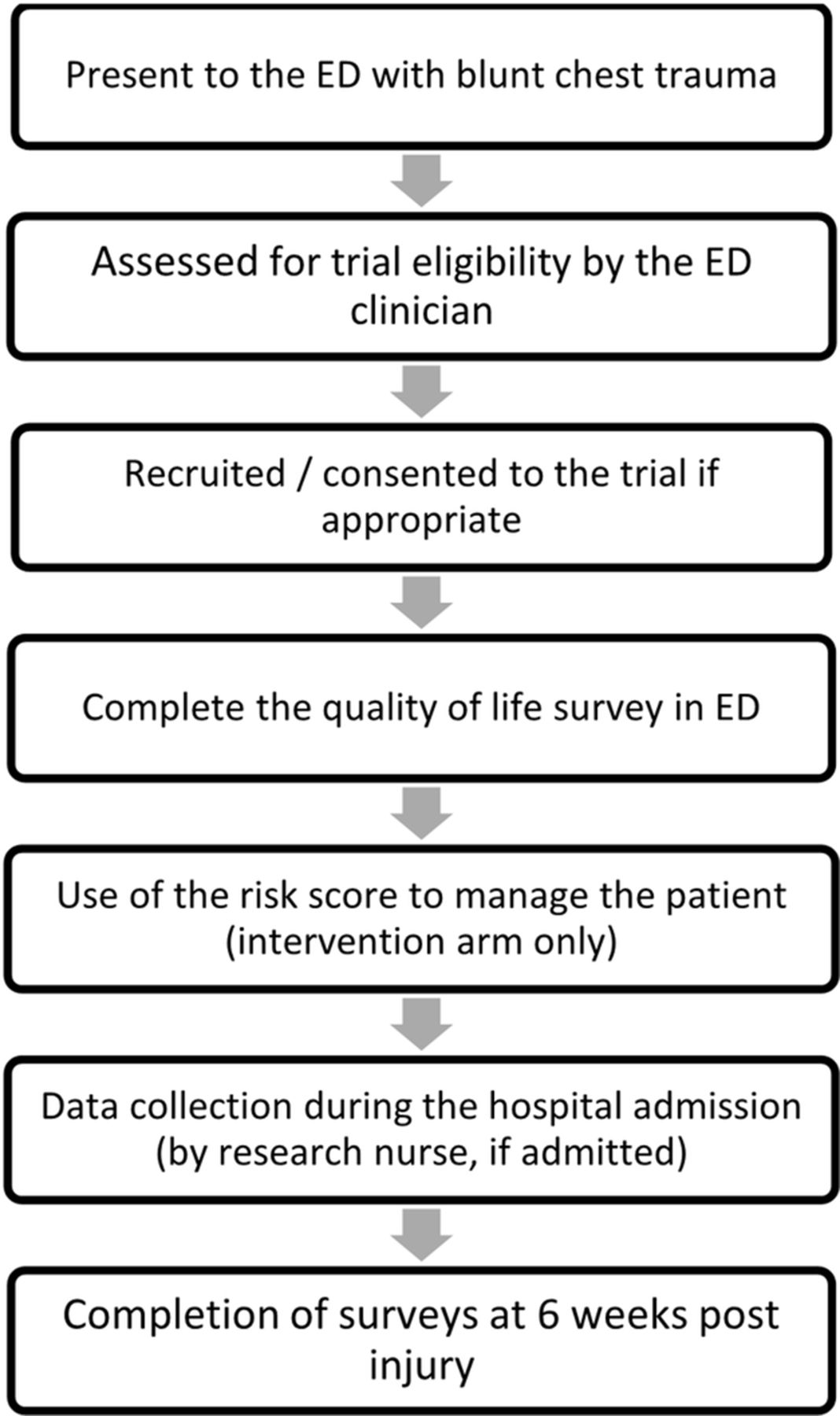
The intervention being investigated in this study is the use of the prognostic model to guide the clinicians' clinical decision-making. The model will be used by the clinician during the initial patient assessment and will provide a suggestion of the appropriate management in terms of whether the patient can be safely discharged home or whether they need admission to either a ward or critical care. There will be no other differences in patient care. Figure 2 describes the patients’ journey through the trial. All patients will be required to complete one survey (SF-12v2),15 on initial presentation to the ED and two more surveys at 6 weeks post injury (SF-12v2 and a Client Services Receipt Inventory). A sample of the clinicians using the model will be asked to attend a short interview in which they will be asked to discuss the model in relation to clinical practice. The research nurses will be required to attend a focus group in which there will be a discussion about the trial’s methodology including specifically issues around data collection.
![[SP1.jpg]](https://bmjopen.bmj.com/content/bmjopen/7/7/e015972/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
{kind=link}
{kind=link}
Summary of the patients’ journey through trial. ED, emergency department.
Strategies to improve adherence to interventions
Full training (including a training manual) on the use of the model and the trial design will be provided for each hospital’s PI, who will then be responsible for training the doctors working in their ED. Feedback from a sample of the clinicians regarding the model will be provided through a short interview, at a convenient time and location. All documentation will be available in the Welsh language where applicable, working in conjunction with the Language Awareness Infrastructure Support Service. Patients will be offered the chance to be entered into a prize draw if they return their surveys at 6 weeks post injury.
Outcome measures
Primary outcome measure
Patient recruitment rate will be recorded as the number of eligible patients who consent to trial participation at the end of the 5-month data collection period, at each site.
Secondary outcome measures
‘Clinician recruitment rate’ will be recorded as the number of eligible clinicians working within the participating ED who agree to take part in the study by the end of the 5-month data collection period, at each site.
‘Response rate of follow-up data’ (quality-of-life surveys) will be recorded as the number of patients returning postal surveys at the end of the 5-month data collection period, at each site.
‘Clinicians Training Attendance rate’ will be recorded as the number of clinicians who receive formal training in the use of the model, prior to the second data collection period (intervention arm), at each site.
‘Compliance with use of model rate’ will be recorded as the number of times the clinician used the risk model for an eligible patient, at the end of the second data collection period (intervention arm).
‘Overall mean quality of life’ as reported using the SF-12v2 survey for patients, at the end of the control and intervention data collection periods (compared with baseline taken on initial assessment in the ED), at each participating site.
‘Resource usage by patients as a result of the intervention’ as reported on the Client Services Receipt Inventory at 6-week follow-up.
Data collection
Data collection to be completed by the on-site research nurse and will include (1) SF-12v2 survey and prognostic model (if used) completed in the ED; (2) mechanism of injury, a measure of pre-admission frailty, admission status, hospital and intensive care unit (ICU) length of stay, occurrence of complications (mortality or pulmonary morbidity) and readmission to hospital will be obtained from medical records prospectively, during the patient's hospitalisation; and (3) at 6 weeks post injury, the second quality-of-life questionnaire (SF-12) and Client Services Receipt Inventory will be sent out to the patient for completion.
Data collection to be completed by the qualitative researcher will include (1) attendance at the PI-led training sessions at two sites in order to assess consistency of training provided between sites and the appropriateness of the training manual; (2) clinicians will be asked to complete an online feedback survey after the training session to collect their views about the training, for example; travel and time costs incurred, completeness, appropriateness and if they have any suggestions about possible improvements; (3) a total of eight doctors (two from each site) who are using the model will be interviewed at the end of the study period to understand how the tool was used, ease and time of use, as well as problems with its use. If it arises, the study team will need to also understand a situation in which the model could have been used, but was not used, and why that might have occurred. Doctors at participating sites who have not used the model (if any) will be invited to participate in interviews to explore their reasons; and (4) a focus group will be facilitated by the qualitative researcher in which the research nurses responsible for data collection will discuss the trial process in depth.
An evaluation of the feasibility of health economics outcomes data will be completed as part of the trial. As this is a feasibility trial, the focus will be on establishing the most appropriate framework for a future health economic analysis in the full definitive trial. The feasibility of collecting data on outcome and resource use will be assessed. A provisional assessment of the cost categories associated with the intervention (eg, staff time associated with training and use of the model) will be completed through discussion with the qualitative researcher completing the telephone interviews. To capture resource usage by patients as a result of the intervention, from a National Health Service/Personal Social Service perspective, an adapted resource usage questionnaire will be used. The use of the SF-12v2 will be considered as a measure to derive utilities using the SF-6D. Data will be assessed to examine the completeness of data captured, such as response rate and potential missing items.
Data management
Research Electronic Data Capture (REDCap) will be used for data capture and for completion of the electronic case report forms, hosted at Swansea University.16 REDCap is a secure, web-based application designed to support data capture for research studies, providing (1) an intuitive interface for validated data entry, (2) audit trails for tracking data manipulation and export procedures, (3) automated export procedures for seamless data downloads to common statistical packages and (4) procedures for importing data from external sources. The use of the REDCap system will be discussed in the focus group with the research nurses who will be completing the electronic case report forms. Feedback will be used to adapt the system to improve the setup, prior to the commencement of the full definitive trial.
Statistical methods
Quantitative data analysis
Criteria of study success: In order to evaluate the success of the feasibility trial, with the view to continue to a full definitive impact trial, the Data Monitoring Committee will assess the final results using the ACCEPT model, developed by Charlesworth et al).17 The predetermined success criteria are highlighted in box 1.
Trial feasibility criteria
Sample size and participants
≥95% of clinicians working within the participating emergency department agree to take part in the study.
≥80% of eligible patients consent to data collection and follow-up.
Follow-up data for primary outcomes can be collected for ≥80% of patients.
Interventions
All clinicians involved in the trial receive formal training in the use of the model.
90% compliance with use of the model during intervention period.
Outcomes
Mean quality of life reported in the intervention arm is not less than 80% of that reported in the control arm.
Outcome measures reported in the intervention period are equal to, or better than, those reported during the conventional management period.
Qualitative data analysis
Focus groups and interviews will be audio recorded and sent to a professional transcription service for verbatim transcription. The qualitative researcher will oversee this process and on receipt of transcripts will check them for accuracy against the original recording and undertake anonymisation in accordance with best practice standards. The cleaned and anonymised transcripts will be uploaded into NVIVO 10—a qualitative data analysis program.18 The qualitative researcher will code the transcripts thematically using a code book which developed initially from the background literature and feedback on the training as well as issues which will emerge through the process of constant comparison which underpins qualitative data analysis. A subsample of coded transcripts will be checked by a second qualitative researcher (by reciprocal arrangement within the wider team at the trials unit) using the coder comparison query tool.
Trial monitoring and managemen
An independent, joint Trial Steering/Data Monitoring Committee will be formed that has no link to the sponsor and has no competing interests. The role of the committee will be to monitor adverse and serious adverse events, stopping criteria and trial endpoint success criteria analysis. Stopping criteria will include (1) a 5% increase in each of the hospitals, in the number of patients with an unplanned representation to the ED due to development of complications, leading to I admission and unexpected death (following direct discharge from the ED on initial presentation), in the intervention period compared with the control period; and (2) a 5% increase in each of the hospitals, in the number of patients identified as having a delayed admission to ICU leading to unexpected death, in the intervention period compared with the control period. The committee will also be responsible for the overall supervision of the trial to ensure the trial is completed according to rigorous sta
There will be no direct change to patient care; however, once the model has been introduced to each ED, the clinicians will be required to use the prognostic model to guide their management decisions for the patient. The model will only be used as a guide to management; however, if the clinician believes the suggestion of the risk score places the patient at risk, they can over-ride the prognostic model. This would be documented and reviewed by the trial team. Although there are a number of expected adverse events for blunt chest wall trauma patients, the PI may take appropriate urgent safety measures in order to protect research participants against any immediate hazard to their health or safety, without prior authorisation from a regulatory body. Any serious adverse event and urgent safety measures will be reported to the CI immediately with details of the measure and a plan for further action. The chief investigator (CI) or sponsor will notify the main Research Ethics Committee and Trial Steering Committee. Serious adverse events will include (1) death or ICU admission in patients who have been discharged home as suggested by the risk score and (2) any trial patient complaint.
Ethics and dissemination
Ethical issues
This trial has received ethics approval by the Wales Research Ethics Committee 6 (ref: 16/WA/0290). Any arising important protocol modifications (such as changes to eligibility criteria, outcomes, analyses) will be communicated to the relevant parties (investigators, Research Ethics Committee / International Review Board, trial participants, trial registries, journals, regulators) in a timely manner. Compliance with this will be monitored by the trial sponsor (ABMU Health Board R&D Department). Informed consent will be obtained by the clinicians or research nurses who will all have received ‘protocol and informed consent specific training’ in alignment with the principles of Good Clinical Practice and who have signed the trial delegation log. Consent will be sought, following a full introduction to the study and once the patient has had time to discuss the Patient Information Sheet with a family member/carer (as appropriate). A study withdrawal letter will also be attached to the Patient Information Sheet in case the patient wishes to withdraw consent in the first week following recruitment.
The trial’s CI will take responsibility to ensure that patient anonymity is protected and maintained. Information with regards to study patients will be kept confidential and managed in accordance with the Data Protection Act, NHS Caldicott Guardian, The Research Governance Framework for Health and Social Care and Research Ethics Committee Approval. All patients will be allocated a study number once informed consent is obtained. Personal data will only be identifiable by this study number during data collection. All patient identifiable data will be removed and data anonymised once data collection using the survey is complete. The CI will act as the custodian of the data and the records will be kept securely for a further 5 years in the Health Board archive facility. The Caldicott Guidelines will be adhered to throughout the study.
Dissemination policy
Dissemination of the outputs from this trial is proposed through publication in an appropriate Emergency Medicine Journal and by presentation at relevant international conferences. The aim of this feasibility trial is not to report definitive results regarding clinical and cost effectiveness; however, any important outputs produced in the trial related to the prognostic model will be published in appropriate medical journals as follow-on articles from our previous published work in this area. We will disseminate our findings to stakeholders via professional meetings. The Trauma and Audit Research Network (TARN) newsletter will be used to disseminate the results to the Trauma leads in each ED participating in TARN in the UK.
References
Footnotes
Contributors All authors of the paper have contributed to the design of the trial. CB wrote this protocol and all other authors edited and made revisions for intellectual content. PE, HH FL, JG and SJ have been involved in the background development and validation work leading up to this trial. For the protocol development, PE, FL, RB, SJ and JG provided the Emergency Medicine expertise, SG provided the health economic expertise, AW provided the statistical expertise, HH provided patient reported outcomes expertise, HH and ZA and overall methodological expertise and CON developed and wrote the qualitative aspects of the protocol. All authors have read and approved the final manuscript for publication.
Funding This trial is supported by a Research for Patient and Public Benefit (RfPPB) Grant by Health and Care Research Wales. Project reference: 1193.
Disclaimer The funding sources have no role in the design of this trial. The views expressed are those of the author(s) and not necessarily those of the NHS, Health and Care Research Wales, the NIHR or the Department of Health
Competing interests None declared.
Ethics approval Wales REC 6.
Provenance and peer review Not commissioned; externally peer reviewed.