Synthesis and Biological Evaluation of New Antitubulin Agents Containing 2-(3′,4′,5′-trimethoxyanilino)-3,6-disubstituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine Scaffold

 , , , and
, , , and
Abstract
:
1. Introduction
2. Chemistry
3. Results and Discussion
3.1. In Vitro Antiproliferative Activities Against A Panel of Three Different Cancer Cell Lines (L1210, CEM, and HeLa)
3.2. Inhibition of Tubulin Polymerization and Colchicine Binding
3.3. Analysis of Effects on the Cell Cycle
3.4. Effects on Apoptosis
3.5. Molecular Modeling Studies
4. Materials and Methods
4.1. Chemistry
4.1.1. Materials and Methods
4.1.2. General Procedure A for the Synthesis of Compounds (4a–4p)
4.1.3. General Procedure B for the Synthesis of Compounds (5a–p)
4.1.4. General Procedure C for the Preparation of Compounds (3a–3p)
4.2. Biological Evaluation
4.2.1. Cell Growth Conditions and Antiproliferative Assay
4.2.2. Effects on Tubulin Polymerization
4.2.3. Effects on the Cell Cycle
4.2.4. Effects on Apoptosis
4.2.5. Molecular Modeling Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Center, M.M.; De Sanis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worlwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Akhdar, H.; Legendre, C.; Aninat, C.; Morel, F. Anticancer drug metabolism: Chemotherapy resistance and new therapeutic approaches. In Topics on Drug Metabolism; Paxton, J., Ed.; InTech: Rijeka, Croatia, 2012; pp. 137–170. [Google Scholar]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Narang, A.S.; Desai, D.S. Anticancer drug development: Unique aspects of pharmaceutical development. In Pharmaceutical Perspectives of Cancer Therapeutics; Lu, Y., Mahato, R.I., Eds.; Springer Science: New York, NY, USA, 2009; pp. 49–92. [Google Scholar]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [Green Version]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, G.; Gill, R.K.; Soni, R.; Bariwal, J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. [Google Scholar] [CrossRef]
- van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Nitika, V.; Kapil, K. Microtubule targeting agents: A benchmark in cancer therapy. Curr. Drug Ther. 2014, 8, 189–196. [Google Scholar] [CrossRef]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- Seligmann, J.; Twelves, C. Tubulin: An example of targeted chemotherapy. Future Med. Chem. 2013, 5, 339–352. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Dráber, P. Tubulins as therapeutic targets in cancer: From bench to bedside. Curr. Pharm. Des. 2012, 18, 2778–2792. [Google Scholar] [CrossRef] [PubMed]
- Manka, S.W.; Moores, C.A. The role of tubulin–tubulin lattice contacts in the mechanism of microtubule dynamic instability. Nat. Struct. Mol. Biol. 2018, 25, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benarroch, E.E. Dynamics of microtubules and their associated proteins: Recent insights and clinical implications. Neurology 2016, 86, 1911–1920. [Google Scholar] [CrossRef]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef]
- Manfredi, J.; Parness, J.; Horwitz, S. Taxol binds to cellular microtubules. J. Cell Biology 1982, 94, 688–696. [Google Scholar] [CrossRef] [Green Version]
- Moudi, M.; Go, R.; Yien, C.; Nazre, M. Vinca alkaloids. Int. J. Prev. Medicine. 2013, 4, 1231–1235. [Google Scholar]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef] [Green Version]
- Skoufias, D.; Wilson, L. Mechanism of inhibition of microtubule polymerization by colchicine: Inhibitory potencies of unliganded colchicine and tubulin-colchicine complexes. Biochemistry 1992, 31, 738–746. [Google Scholar] [CrossRef]
- Garon, E.B.; Neidhart, J.D.; Gabrail, N.Y.; de Oliveira, M.R.; Balkissoon, J.; Kabbinavar, F. A randomized Phase II trial of the tumor vascular disrupting agent CA-4P (fosbretabulin tromethamine) with carboplatin, paclitaxel, and bevacizumab in advanced nonsquamous non-small-cell lung cancer. Onco Targets Ther. 2016, 9, 7275–7283. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than just vascular targeting agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Vindya, N.G.; Sharma, N.; Yadav, M.; Ethiraj, K.R. Tubulins-the target for anticancer therapy. Curr. Top. Med. Chem. 2015, 15, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.N.; Zheng, L.L.; Wang, D.; Liang, X.X.; Gao, F.; Zhou, X.L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmaol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Theeramunkong, S.; Caldarelli, A.; Massarotti, A.; Aprile, S.; Caprifoglio, D.; Zaninetti, R.; Teruggi, A.; Pirali, T.; Grosa, G.; Tron, G.C.; et al. Regioselective Suzuki coupling of dihaloheteroaromatic compounds as a rapid strategy to synthesize potent rigid combretastatin analogues. J. Med. Chem. 2011, 54, 4977–4986. [Google Scholar] [CrossRef]
- Flynn, B.L.; Flynn, G.P.; Hamel, E.; Jung, M.K. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin A–4. Bioorg. Med. Chem. Lett. 2001, 11, 2341–2343. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Q.; Bai, Z.; Sun, J.; Jiang, X.; Song, H.; Wub, Y.; Zhang, W. Synthesis and biological evaluation of 2,3–diarylthiophene analogues of combretastatin A-4. Med. Chem. Commun. 2015, 6, 971–976. [Google Scholar] [CrossRef]
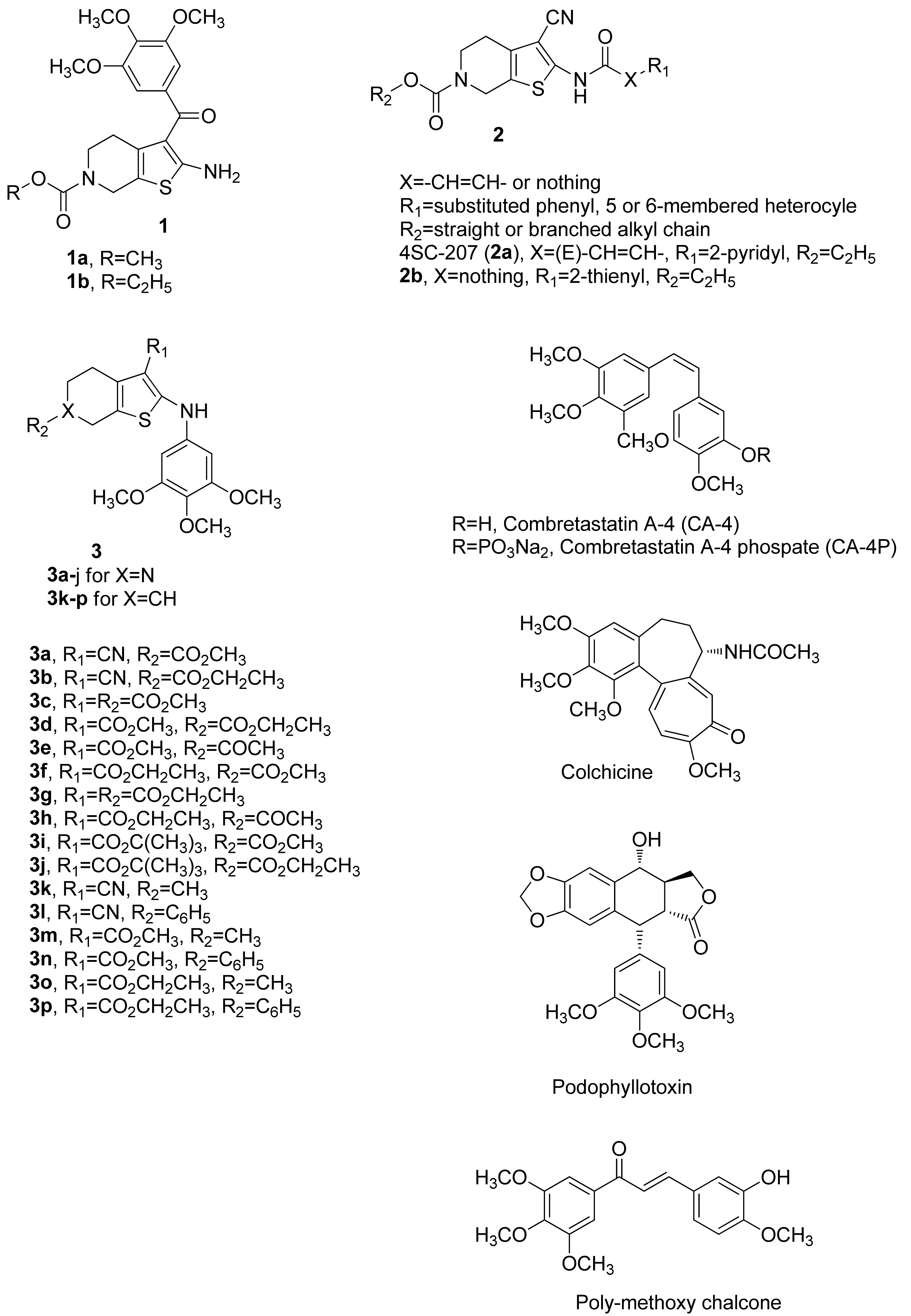
- Romagnoli, R.; Baraldi, P.G.; Remusat, V.; Carrion, M.D.; Lopez Cara, C.; Preti, D.; Fruttarolo, F.; Pavani, M.G.; Aghazadeh Tabrizi, M.; Tolomeo, M.; et al. Synthesis and biological evaluation of 2-(3′,4′,5′–trimethoxybenzoyl)-3-amino 5-aryl thiophenes as a new class of tubulin inhibitors. J. Med. Chem. 2006, 49, 6425–6428. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Pavani, M.G.; Tabrizi, M.A.; Preti, D.; Fruttarolo, F.; Piccagli, L.; Jung, M.K.; Hamel, E.; Borgatti, M.; et al. Synthesis and biological evaluation of 2-amino-3-(3′, 4′, 5′-trimethoxybenzoyl)-5-aryl thiophenes as a new class of potent antitubulin agents. J. Med. Chem. 2006, 49, 3906–3915. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Balzarini, J.; Nussbaumer, P.; Bassetto, M.; Brancale, A.; et al. Design, synthesis and biological evaluation of 3,5-disubstituted 2-amino thiophene derivatives as a novel class of antitumor agents. Bioorg. Med. Chem. 2014, 22, 5097–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Somappa, S.B.; Patil, S.A.; Nagaraja, B.M. An overview of benzo[b]thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017, 138, 1002–1033. [Google Scholar] [CrossRef] [PubMed]
- Pinney, K.G.; Bounds, A.D.; Dingerman, K.M.; Mocharla, V.P.; Pettit, G.R.; Bai, R.; Hamel, E. A new anti-tubulin agent containing the benzo[b]thiophene ring system. Bioorg. Med. Chem. Lett. 1999, 9, 1081–1086. [Google Scholar] [CrossRef]
- Flynn, B.L.; Verdier-Pinard, P.; Hamel, E. A novel palladium-mediated coupling approach to 2,3-disubstituted benzo[b]thiophenes and its application to the synthesis of tubulin binding agents. Org. Lett. 2001, 3, 651–654. [Google Scholar] [CrossRef]
- Chen, Z.; Mocharla, V.P.; Farmer, J.M.; Pettit, G.R.; Hamel, E.; Pinney, K.G. Preparation of new anti-tubulin ligands through a dual-mode, addition-elimination reaction to a bromo-substituted α, β-unsaturated sulfoxide. J. Org. Chem. 2000, 65, 8811–8815. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Lopez Cara, C.; Hamel, E.; Basso, G.; Bortolozzi, R.; Viola, G. Synthesis and biological evaluation of 2-(3′,4′,5′-trimethoxybenzoyl)-3-aryl/arylaminobenzo[b]thiophene derivatives as a novel class of antiproliferative agents. Eur. J. Med. Chem. 2010, 45, 5781–5791. [Google Scholar] [CrossRef] [Green Version]
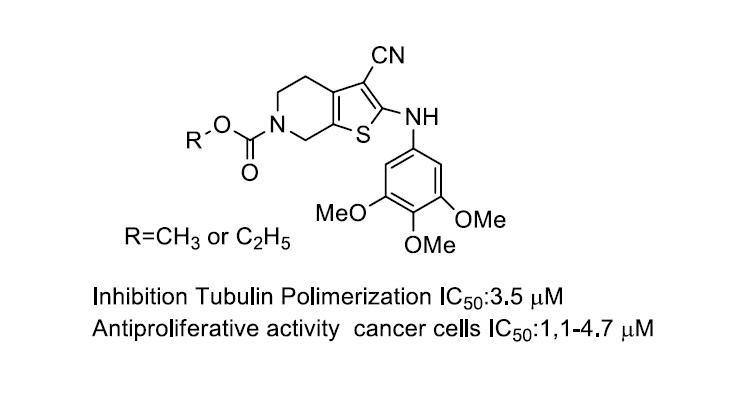
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Bassetto, M.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Bortolozzi, R.; et al. Synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilino benzo[b]thiophenes and thieno[2,3-c]pyridines as new potent anticancer agents. J. Med. Chem. 2013, 56, 2606–2618. [Google Scholar] [CrossRef] [Green Version]
- Kemnitzer, W.; Sirisoma, N.; May, C.; Tseng, B.; Drewe, J.; Xiong Cai, S. Discovery of 4-anilino-N-methylthieno[3,2-d]pyrimidines and 4-anilino-N-methylthieno[2,3-d]pyrimidines as potent apoptosis inducers. Bioorg. Med. Chem. Lett. 2009, 19, 3536–3540. [Google Scholar] [CrossRef]
- Tian, C.; Chen, X.; Zhang, Z.; Wang, X.; Liu, J. Design and synthesis of (2-(phenylamino)thieno[3,2-d]pyrimidin-4-yl)(3,4,5-trimethoxyphenyl)methanone analogues as potent antitubulin polymerization agents. Eur. J. Med. Chem. 2019, 183, 111679. [Google Scholar] [CrossRef]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Baraldi, S.; Baraldi, P.G.; Schiaffino Ortega, S.; Chayah, M.; Kimatrai Salvador, M.; Lopez-Cara, L.C.; Brancale, A.; et al. Design, synthesis and biological evaluation of 6-substituted thieno[3,2-d]pyrimidine analogues as dual epidermal growth factor receptor kinase and microtubule inhibitors. J. Med. Chem. 2019, 62, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- Eurtivong, C.; Semenov, V.; Semenova, M.; Konyushkin, L.; Atamanenko, O.; Reynisson, J.; Kiselyov, A. 3-Amino-thieno[2,3-b]pyridines as microtubule-destabilising agents: Molecular modelling and biological evaluation in the sea urchin embryo and human cancer cells. Bioorg. Med. Chem. 2017, 25, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Lopez Cara, C.; Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Pipitone, M.R.; Balzarini, J.; et al. Synthesis and biological evaluation of 2-amino-3-(3′,4′,5′-trimethoxybenzoyl)-6-substituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivatives as antimitotic agents and inhibitors of tubulin polymerization. Bioorg. Med. Chem. Lett. 2008, 18, 5041–5045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, B.; Gimmnich, P.; Pekari, K.; Baer, T.; Schmidt, M.; Beckers, T. Tetrahydropyridothiophenes for use in the treatment of cancer. WO2005118592, 15 December 2005. [Google Scholar]
- Pekari, K.; Baer, T.; Bertels, B.; Schmidt, M.; Beckers, T. Novel tetrahydropyridothiophenes. WO2005118071, 15 December 2005. [Google Scholar]
- Bausch, E.; Kohlhof, H.; Hamm, S.; Krauss, R.; Baumgartner, R.; Sironi, L. A novel microtubule inhibitor 4SC-207 with anti-proliferative activity in taxane-resistant cells. PLoS ONE 2013, 8, e79594. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef]
- Wang, K.; Kim, D.; Domling, A. Cyanoacetamide MCR (III): Three-component Gewald reactions revisited. J. Comb. Chem. 2010, 12, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Doyle, M.P.; Siegfried, B.; Dellaria, J.F., Jr. Alkyl nitrite-metal halide deamination reactions. 2. Substitutive deamination of arylamines by alkyl nitrites and copper (II) halides. A direct and remarkably efficient conversion of arylamines to aryl halides. J. Org. Chem. 1977, 42, 2426–2431. [Google Scholar] [CrossRef]
- Queiroz, M.-J.R.P.; Begouin, A.; Ferreira, I.C.F.R.; Kirsch, G.; Calhelha, R.C.; Barbosa, S.; Estevinho, L.M. Palladium-catalysed amination of electron-deficient or relatively electron-rich benzo[b]thienyl bromides-Preliminary studies of antimicrobial activity and SARs. Eur. J. Org. Chem. 2004, 3679–3685. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2019-3: Maestro, Schrödinger, LLC: New York, NY, USA, 2019. Available online: https://www.chemcomp.com (accessed on 7 April 2020).
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargstem, K.; Cavalli, A.; Steinmetz, M.O. Structural basis of cis- and trans-Combretastatin binding to tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Kimatrai Salvador, M.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilinoindoles as a new class of potent inhibitors of tubulin polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E.; Lin, C.M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–21. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Lai, J.-Y.; Yoo, H.-D.; Yu, J.; Marquez, B.; Nagle, D.G.; Nambu, M.; White, J.D.; Falck, J.R.; Gerwick, W.H.; et al. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998, 53, 62–67. [Google Scholar] [CrossRef]
- Milani, R.; Brognara, E.; Fabbri, E.; Manicardi, A.; Corradini, R.; Finotti, A.; Gasparello, J.; Borgatti, M.; Cosenza, L.C.; Lampronti, I.; et al. Targeting miR-155-5p and miR-221-3p by peptide nucleic acids induces caspase-3 activation and apoptosis in temozolomide-resistant T98G glioma cells. Int. J. Oncol. 2019, 55, 59–68. [Google Scholar] [CrossRef] [Green Version]
- ULC, C.C.G. Molecular Operating Environment (MOE), 2019.10, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2019. Available online: https://www.schrodinger.com (accessed on 7 April 2020).
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) a | |||
|---|---|---|---|
| Compound | L1210 | CEM | HeLa |
| 3a | 4.7 ± 0.4 | 2.9 ± 2.1 | 1.9 ± 1.2 |
| 3b | 2.8 ± 2.3 | 2.3 ± 1.7 | 1.1 ± 0.0 |
| 3c | 17 ± 5 | 16 ± 4 | 23 ± 10 |
| 3d | 15 ± 2 | 12 ± 4 | 16 ± 3 |
| 3e | 26 ± 2 | 43 ± 12 | 48 ± 17 |
| 3f | 22 ± 4 | 22 ± 8 | 67 ± 20 |
| 3g | 19 ± 0 | 19 ± 1 | 19 ± 4 |
| 3h | 25 ± 3 | 34 ± 9 | 24 ± 3 |
| 3i | 18 ± 3 | 20 ± 2 | 14 ± 1 |
| 3j | 19 ± 3 | 19 ± 2 | 20 ± 4 |
| 3k | >250 | >250 | 202 ± 25 |
| 3l | 92 ± 29 | 58 ± 53 | 124 ± 8 |
| 3m | 82 ± 13 | 45 ± 17 | 11 ± 6 |
| 3n | 101 ± 9 | 102 ± 50 | 48 ± 6 |
| 3o | 86 ± 41 | 78 ± 33 | 66 ± 6 |
| 3p | 112 ± 1 | 88 ± 12 | 169 ± 18 |
| 2b | 6.0 ± 0.3 | 3.7 ± 1.2 | 41 ± 27 |
| CA-4 (nM) | 2.8 ± 1.1 | 1.9 ± 1.1 | 4.1 ± 0.1 |
| Compound | Tubulin assembly a IC50 ± SD (μM) | Colchicine binding b % ± SD |
|---|---|---|
| 3a | 3.8 ± 0.05 | 29 ± 1 |
| 3b | 3.4 ± 0.05 | 31 ± 1 |
| CA-4 | 0.54 ± 0.06 | 98 ± 0.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romagnoli, R.; Prencipe, F.; Oliva, P.; Cacciari, B.; Balzarini, J.; Liekens, S.; Hamel, E.; Brancale, A.; Ferla, S.; Manfredini, S.; et al. Synthesis and Biological Evaluation of New Antitubulin Agents Containing 2-(3′,4′,5′-trimethoxyanilino)-3,6-disubstituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine Scaffold. Molecules 2020, 25, 1690. https://doi.org/10.3390/molecules25071690
Romagnoli R, Prencipe F, Oliva P, Cacciari B, Balzarini J, Liekens S, Hamel E, Brancale A, Ferla S, Manfredini S, et al. Synthesis and Biological Evaluation of New Antitubulin Agents Containing 2-(3′,4′,5′-trimethoxyanilino)-3,6-disubstituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine Scaffold. Molecules. 2020; 25(7):1690. https://doi.org/10.3390/molecules25071690
Chicago/Turabian StyleRomagnoli, Romeo, Filippo Prencipe, Paola Oliva, Barbara Cacciari, Jan Balzarini, Sandra Liekens, Ernest Hamel, Andrea Brancale, Salvatore Ferla, Stefano Manfredini, and et al. 2020. "Synthesis and Biological Evaluation of New Antitubulin Agents Containing 2-(3′,4′,5′-trimethoxyanilino)-3,6-disubstituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine Scaffold" Molecules 25, no. 7: 1690. https://doi.org/10.3390/molecules25071690