
Synthesis and Biological Evaluation of 2-Substituted Benzyl-/Phenylethylamino-4-amino-5-aroylthiazoles as Apoptosis-Inducing Anticancer Agents

 , , and
, , and
Abstract
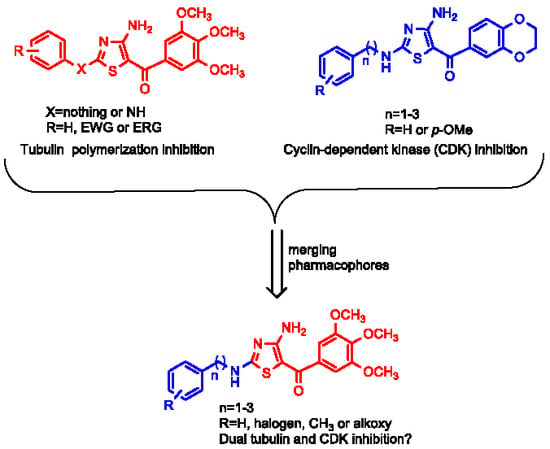
:
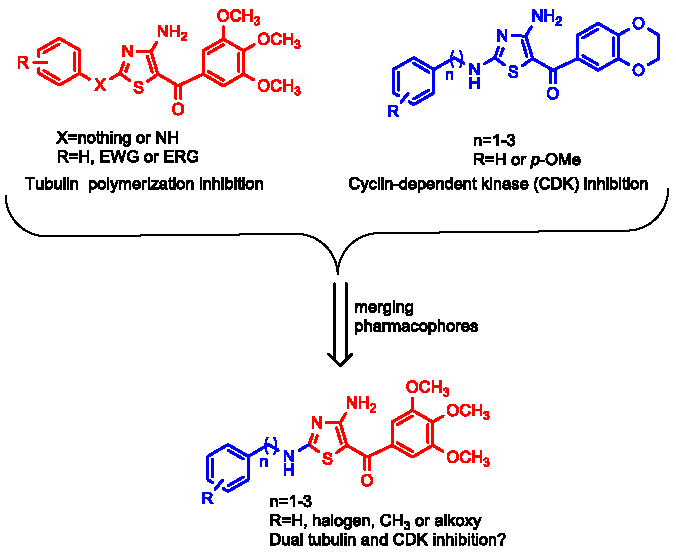
1. Introduction
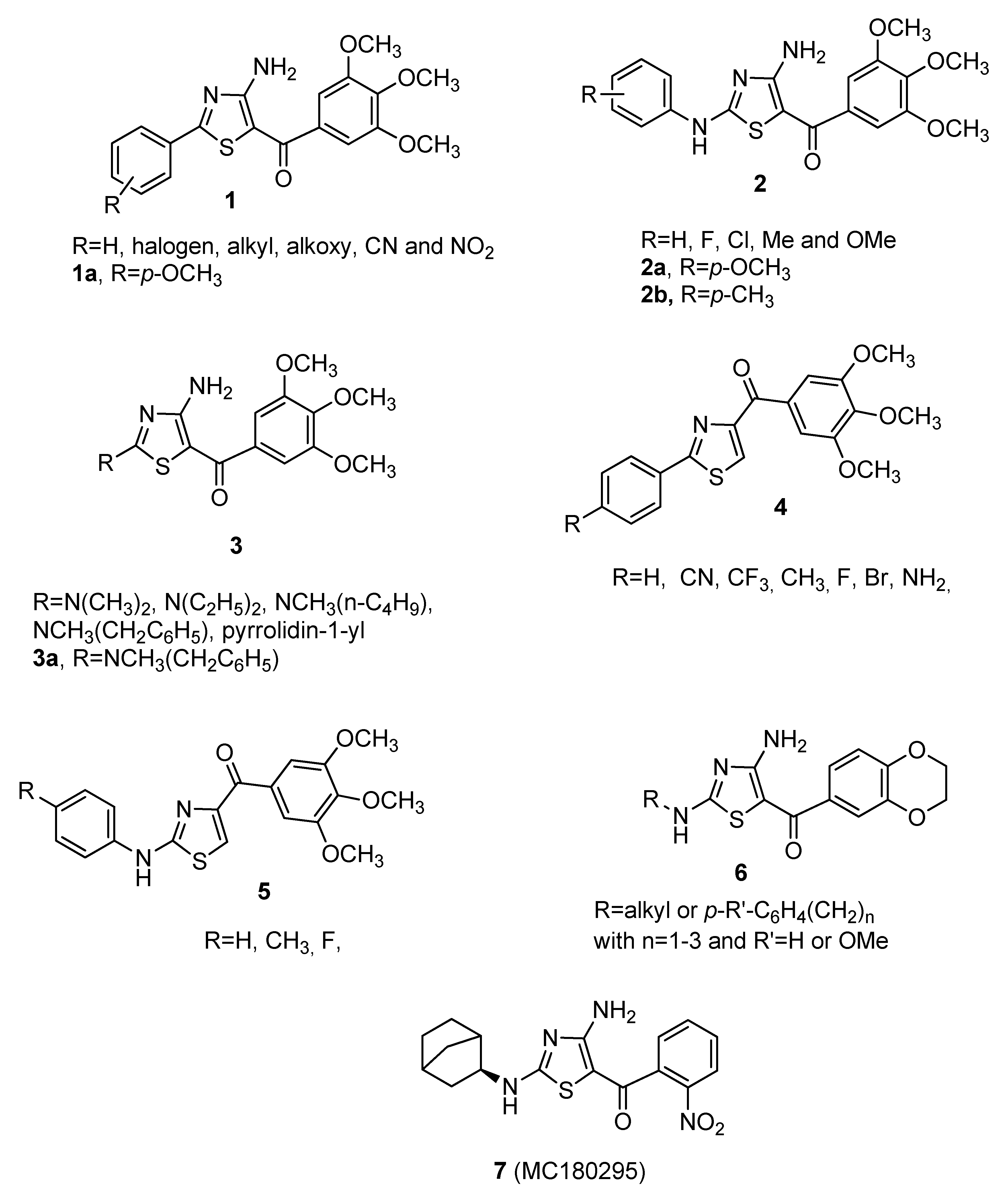
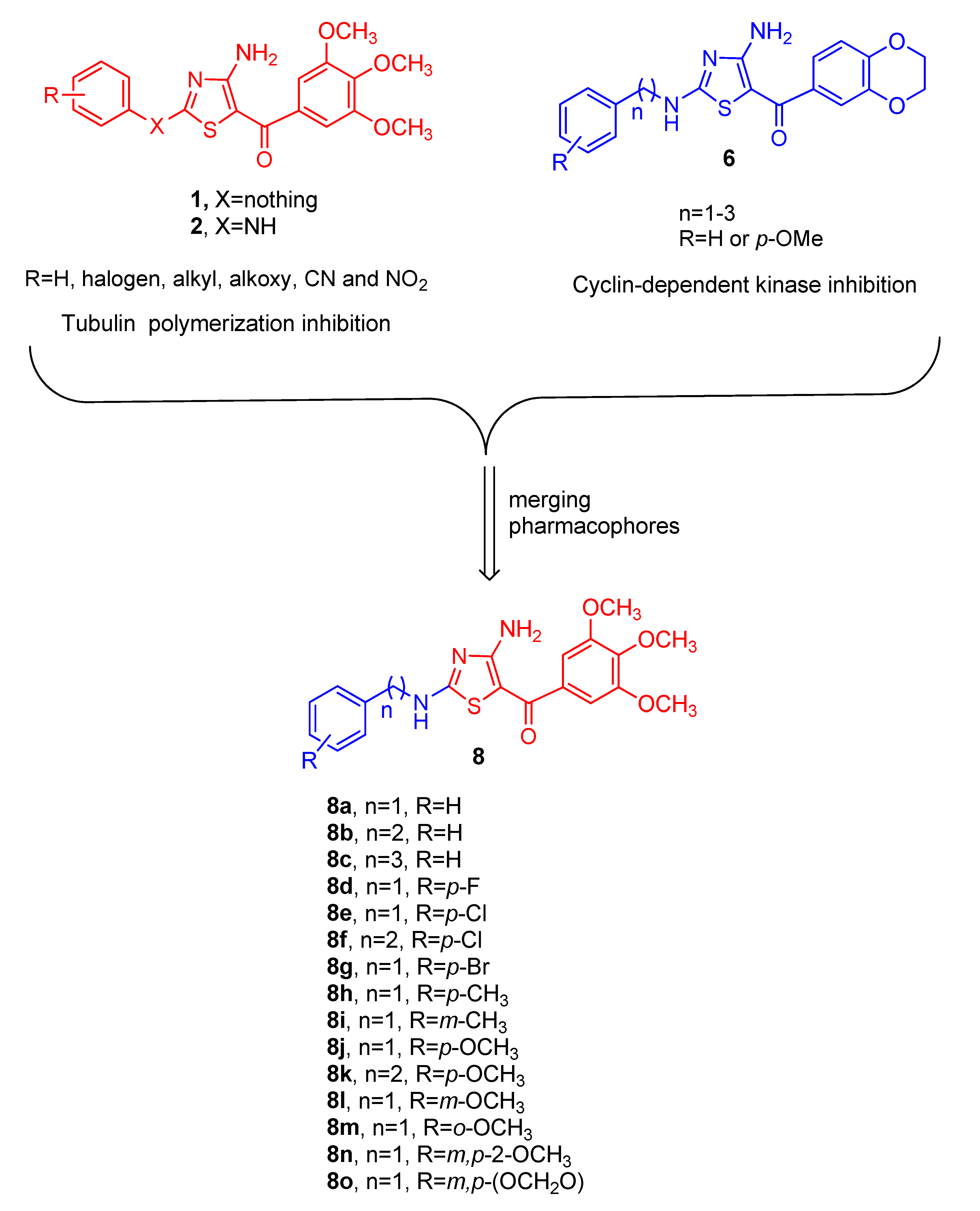
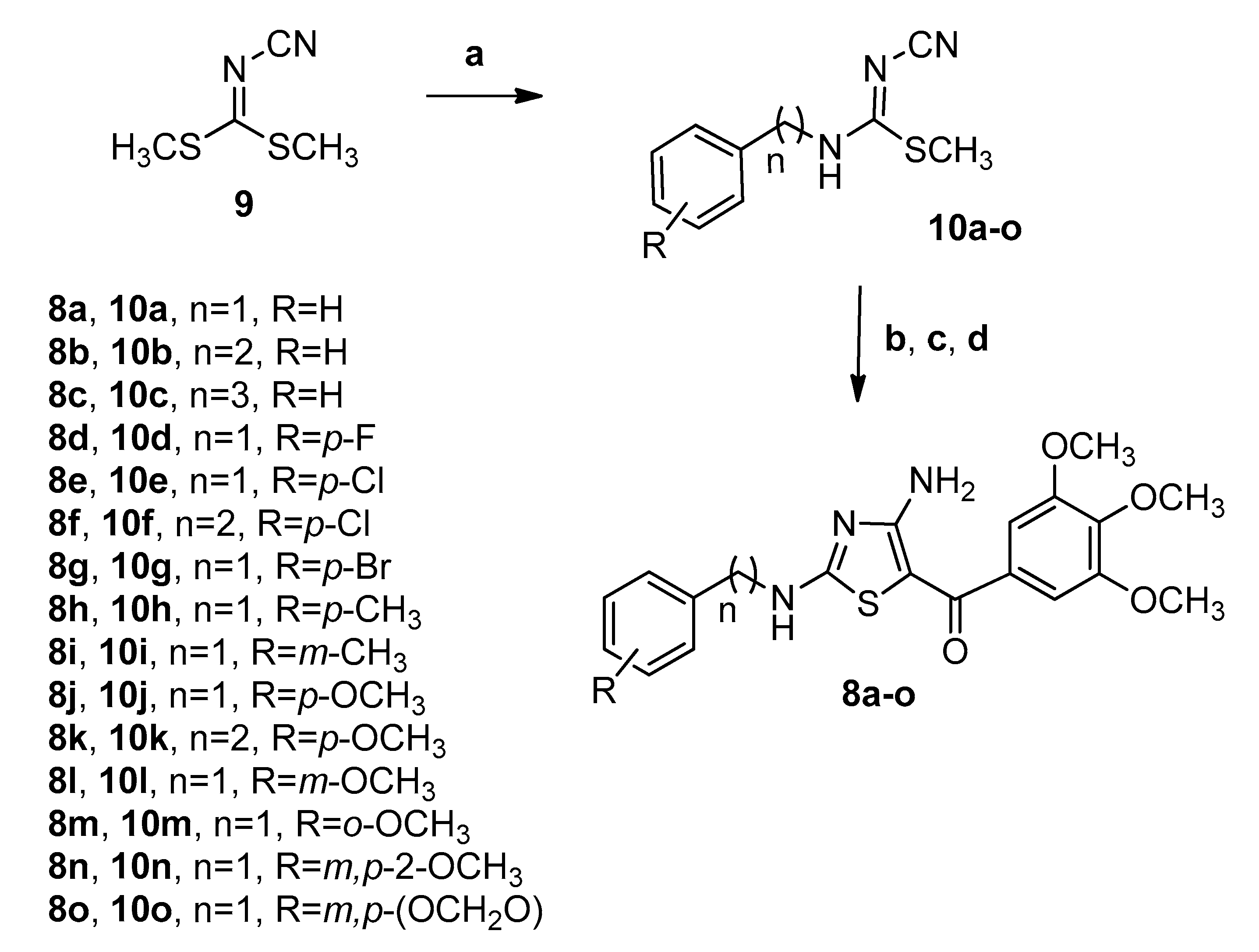
2. Chemistry
3. Results and Discussion
3.1. In Vitro Antiproliferative Activities against Human U-937 and SK-MEL-1 Cell Lines
3.2. Tubulin Polymerization and CDK Inhibitory Activity Assays
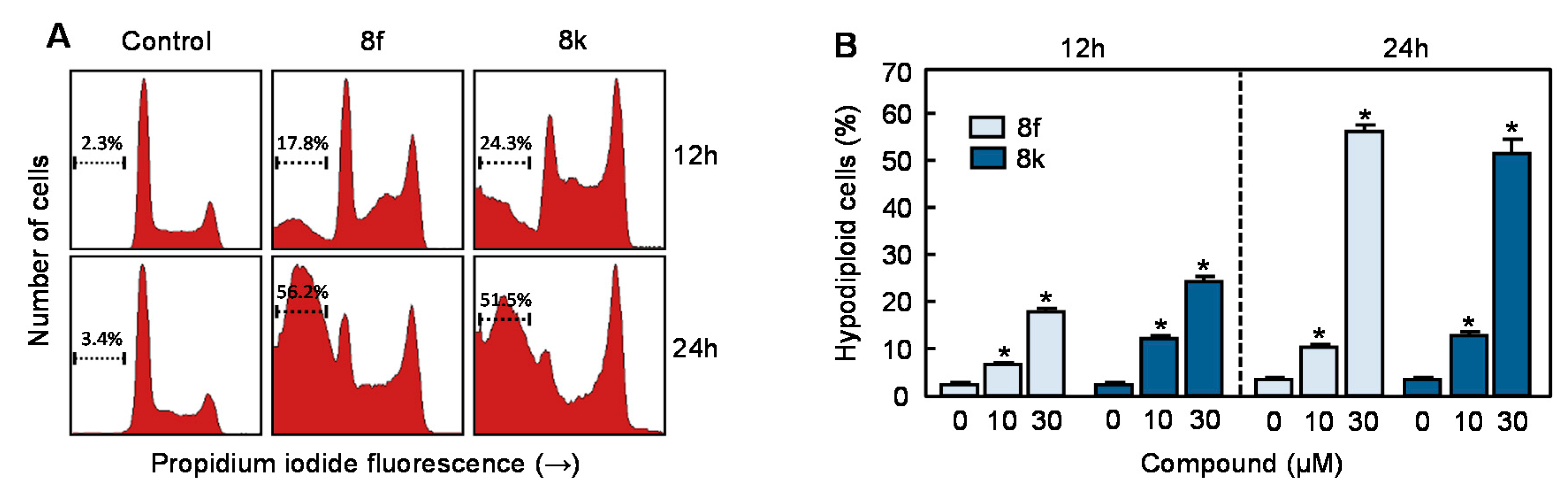
3.3. Flow Cytometric Analysis in Human Myeloid Leukemia Cells
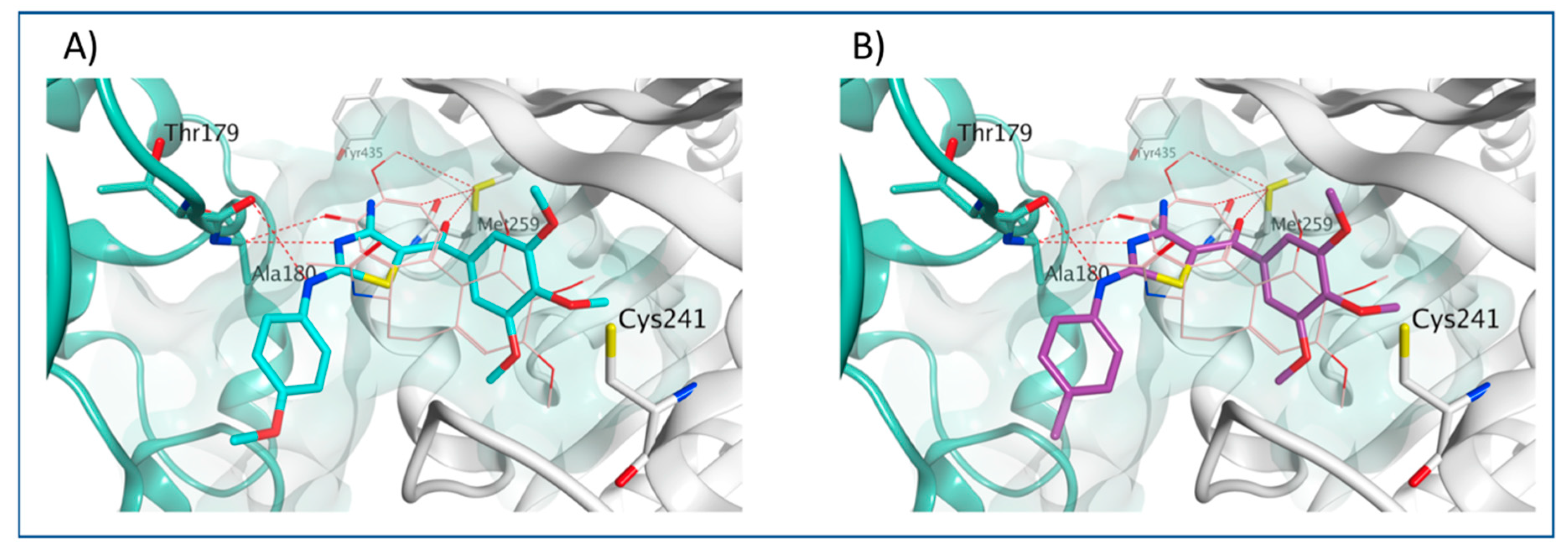
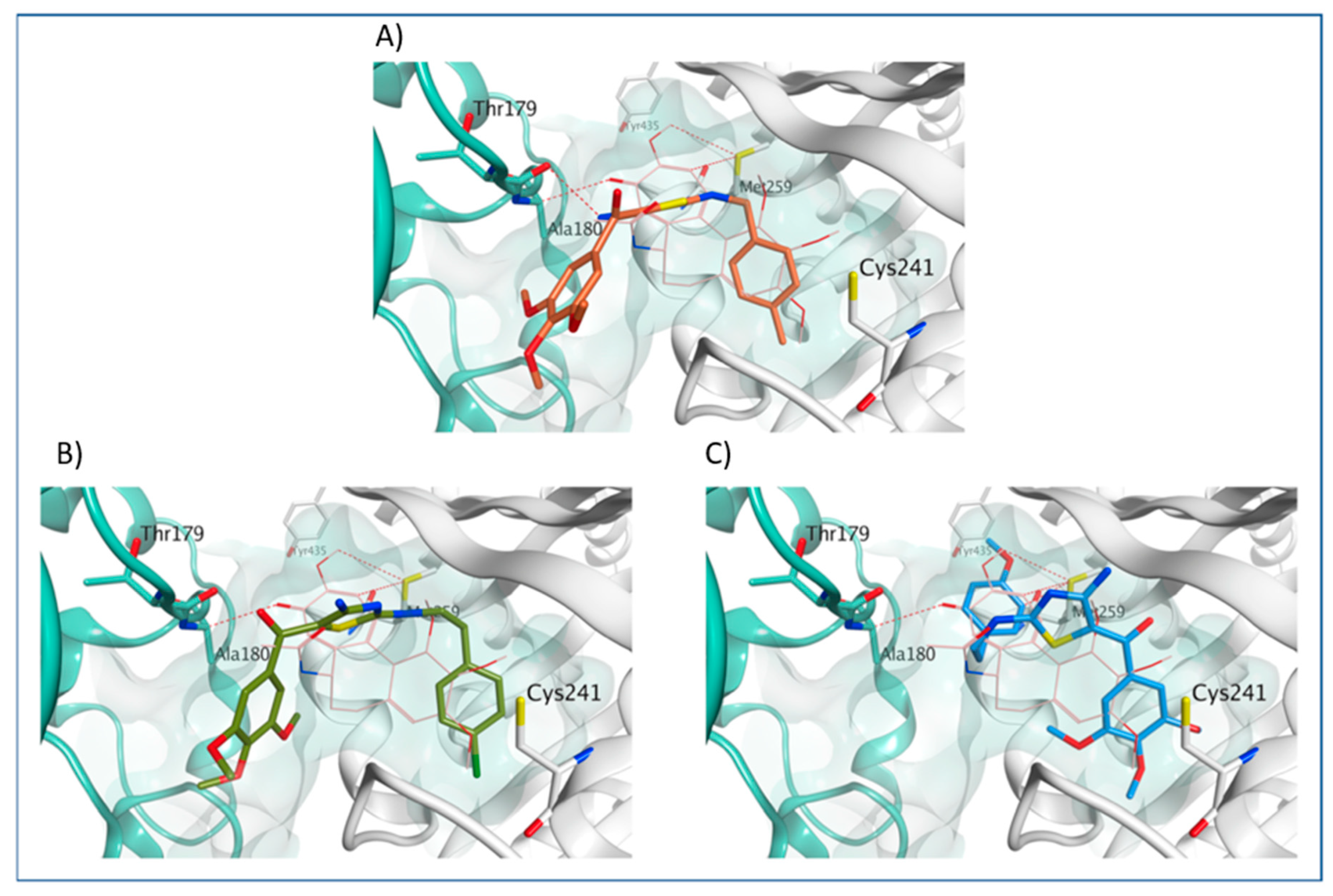
3.4. Molecular Modeling Studies
4. Experimental
4.1. Chemistry
4.1.1. Materials and Methods
4.1.2. General Procedure A for the Synthesis of Compounds 10a–o
(Z)-Methyl N-benzyl-N′-cyanocarbamimidothioate (10a)
(Z)-Methyl N′-cyano-N-phenethylcarbamimidothioate (10b)
(Z)-Methyl N′-cyano-N-(3-phenylpropyl)carbamimidothioate (10c)
(Z)-methyl N-4-fluorobenzyl-N′-cyanocarbamimidothioate (10d)
(Z)-methyl N-4-chlorobenzyl-N′-cyanocarbamimidothioate (10e)
(Z)-Methyl N-4-chlorophenethyl-N′-cyanocarbamimidothioate (10f)
(Z)-methyl N-4-bromobenzyl-N′-cyanocarbamimidothioate (10g)
(Z)-Methyl N′-cyano-N-(4-methylbenzyl)carbamimidothioate (10h)
(Z)-Methyl N′-cyano-N-(3-methylbenzyl)carbamimidothioate (10i)
(Z)-Methyl N′-cyano-N-(4-methoxybenzyl)carbamimidothioate (10j)
(Z)-Methyl N-4-methoxyphenethyl-N′-cyanocarbamimidothioate (10k)
(Z)-Methyl N′-cyano-N-(3-methoxybenzyl)carbamimidothioate (10l)
(Z)-Methyl N′-cyano-N-(2-methoxybenzyl)carbamimidothioate (10m)
(Z)-methyl N′-cyano-N-(3,4-dimethoxybenzyl)carbamimidothioate (10n)
(Z)-Methyl N-(benzo[d][1,3]dioxol-5-ylmethyl)-N′-cyanocarbamimidothioate (10o)
4.1.3. General Procedure B for the Synthesis of Compounds 8a–o
(4-Amino-2-(benzylamino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8a)
(4-Amino-2-(phenethylamino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8b)
(4-Amino-2-((3-phenylpropyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8c)
(4-Amino-2-((4-fluorobenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8d)
(4-Amino-2-((4-chlorobenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8e)
(4-Amino-2-((4-chlorophenethyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8f)
(4-Amino-2-((4-bromobenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8g)
(4-Amino-2-((4-methylbenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8h)
(4-Amino-2-((3-methylbenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8i)
(4-Amino-2-((4-methoxybenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8j)
(4-Amino-2-((4-methoxyphenethyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8k)
(4-Amino-2-((3-methoxybenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8l)
(4-Amino-2-((2-methoxybenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8m)
(4-Amino-2-((3,4-dimethoxybenzyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (8n)
(4-Amino-2-((benzo[d][1,3]dioxol-5-ylmethyl)amino)thiazol-5-yl)(3,4,5-trimethoxyphenyl) methanone (8o)
4.2. Biological Evaluation
4.2.1. Cell Culture and Cell Viability Assays
4.2.2. Analysis of Cell Cycle by Flow Cytometry
4.2.3. Effects on Tubulin Polymerization
4.2.4. In Vitro CDK Inhibition Assay
4.2.5. Molecular Modeling Methods
4.2.6. Statistical Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.S.; Zong, W.-X. Chemotherapeutic Approaches for Targeting Cell Death Pathways. Oncologist 2006, 11, 342–357. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Wang, R.; Gandhi, V. Targeting the apoptosis pathway in hematologic malignancies. Leuk. Lymphoma 2014, 55, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Benarroch, E.E.; Rouleau, G.A. Dynamics of microtubules and their associated proteins: Recent insights and clinical implicationsAuthor Response. Neurology 2016, 86, 1911–1920. [Google Scholar] [CrossRef]
- Pooler, D.B.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2016, 83, 255–268. [Google Scholar] [CrossRef]
- Naaz, F.; Haider, R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Brancale, A.; Fu, X.-H.; Li, J.; Zhang, S.-Z.; Hamel, E.; et al. Discovery and optimization of a series of 2-aryl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl)thiazoles as novel anticancer agents. J. Med. Chem. 2012, 55, 5433–5445. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Lopez-Cara, L.C.; Basso, G.; Viola, G.; Khedr, M.; Balzarini, J.; Mahboobi, S.; et al. 2-Arylamino-4-Amino-5-Aroylthiazoles. “One-Pot” Synthesis and Biological Evaluation of a New Class of Inhibitors of Tubulin Polymerization. J. Med. Chem. 2009, 52, 5551–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Cara, L.C.; Kimatrai Salvador, M.; Bortolozzi, R.; Basso, G.; Viola, G.; Balzarini, J.; Brancale, A.; Fu, X.-H.; et al. One-pot synthesis and biological evaluation of 2-pyrrolidinyl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl)thiazole: A unique, highly active antimicrotubule agent. Eur. J. Med. Chem. 2011, 46, 6015–6024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, C.-M.; Wang, Z.; Ross, C.R.; Chen, J.; Dalton, J.T.; Li, W.; Miller, D.D. Discovery of 4-Substituted Methoxybenzoyl-aryl-thiazole as Novel Anticancer Agents: Synthesis, Biological Evaluation, and Structure−Activity Relationships. J. Med. Chem. 2009, 52, 1701–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, C.-M.; Wang, Z.; Chen, J.; Mohler, M.L.; Li, W.; Dalton, J.T.; Miller, D.D. Design, Synthesis, and SAR Studies of 4-Substituted Methoxylbenzoyl-aryl-thiazoles Analogues as Potent and Orally Bioavailable Anticancer Agents. J. Med. Chem. 2011, 54, 4678–4693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errico, A.; Deshmukh, K.; Tanaka, Y.; Pozniakovsky, A.; Hunt, T. Identification of substrates for cyclin dependent kinases. Adv. Enzym. Regul. 2010, 50, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275. [Google Scholar] [CrossRef]
- Santo, L.; Siu, K.T.; Raje, N. Targeting Cyclin-Dependent Kinases and Cell Cycle Progression in Human Cancers. Semin. Oncol. 2015, 42, 788–800. [Google Scholar] [CrossRef]
- Palanisamy, R.P. Palbociclib: A new hope in the treatment of breast cancer. J. Cancer Res. Ther. 2016, 12, 1220–1223. [Google Scholar] [CrossRef]
- Laderian, B.; Fojo, T. CDK4/6 Inhibition as a therapeutic strategy in breast cancer: Palbociclib, ribociclib, and abemaciclib. Semin. Oncol. 2017, 44, 395–403. [Google Scholar] [CrossRef]
- Issa, J.J.; Zhang, H.; Abu-Gharbia, M.; Childers, W.E.; Morton, G.C. Synthesis of Aminothiazole Compounds for Use in Treatment of Cancer. International Patent Application No. WO2017015484, 26 January 2017. [Google Scholar]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-U. Polypharmacology—Foe or Friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Petrelli, A. From single- to multi-target drugs in cancer therapy: When aspecificity becomes an advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Estévez, S.; Marrero, M.T.; Quintana, J.; Estévez, F. Eupatorin-Induced Cell Death in Human Leukemia Cells Is Dependent on Caspases and Activates the Mitogen-Activated Protein Kinase Pathway. PLoS ONE 2014, 9, e112536. [Google Scholar] [CrossRef] [Green Version]
- Hamel, E.; Lin, C.M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. [Google Scholar] [CrossRef]
- Hamel, E. Evaluation of Antimitotic Agents by Quantitative Comparisons of Their Effects on the Polymerization of Purified Tubulin. Cell Biophys. 2003, 38, 1–22. [Google Scholar] [CrossRef]
- Schordinger Software, Version 8.0. Available online: http://www.schrodinger.com/ (accessed on 10 November 2007).
- Schrödinger Release 2019-3: Maestro, Schrödinger, LLC: New York, NY, USA. 2019. Available online: https://www.chemcomp.com (accessed on 10 April 2020).
Sample Availability: Samples of the compounds 8e, 8f and 8k are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | IC50 (μM) a | |
|---|---|---|
| U-937 | SK-MEL-1 | |
| 8a | 21.1 ± 5.1 | 14.2 ± 9.7 |
| 8b | 19.3 ± 5.6 | 20.5 ± 6.9 |
| 8c | 10.7 ± 2.8 | 16.7 ± 7.0 |
| 8d | 39.1 ± 4.2 | 51.0 ± 1.4 |
| 8e | 8.7 ± 5.4 | 8.3 ± 5.5 |
| 8f | 6.7 ± 3.6 | 12.2 ± 3.2 |
| 8g | 12.9 ± 3.3 | 15.7 ± 4.9 |
| 8h | 15.4 ± 4.9 | 19.0 ± 9.4 |
| 8i | 18.1 ± 6.4 | 16.7 ± 5.6 |
| 8j | 23.8 ± 5.2 | 22.9 ± 13.7 |
| 8k | 5.7 ± 1.3 | 8.0 ± 4.3 |
| 8l | 12.3 ± 6.1 | 12.4 ± 4.8 |
| 8m | 13.6 ± 5.3 | 10.4 ± 2.8 |
| 8n | 28.7 ± 10.5 | 32.7 ± 7.7 |
| 8o | 27.0 ± 0.9 | 24.7 ± 1.0 |
| 1a | 34.3 ± 2.2 | 48.3 ± 4.6 |
| 2a | 0.484 ± 0.368 | 5.2 ± 1.4 |
| CA-4 | 0.019 ± 0.012 | 3.1 ± 2.9 |
| Doxorubicin | 0.083 ± 0.021 | 0.274 ± 0.070 |
| Etoposide | 2.1 ± 0.4 | 6.3 ± 1.0 |
| Time | Compd | µM | % Sub-G1 | % G1 | % S | % G2-M |
|---|---|---|---|---|---|---|
| 6 h | 0 | 4.7 ± 0.2 | 47.3 ± 0.2 | 25.6 ± 0.1 | 20.7 ± 0.8 | |
| 8f | 10 | 4.4 ± 0.5 | 40.4 ± 1.0 * | 29.3 ± 0.1 * | 23.6 ± 1.3 | |
| 30 | 3.8 ± 0.1 | 40.5 ± 1.6 * | 30.9 ± 0.4 * | 23.3 ± 1.4 | ||
| 8k | 10 | 3.7 ± 0.4 | 38.7 ± 0.3 * | 30.7 ± 0.1 * | 25.3 ± 0.2 * | |
| 30 | 7.6 ± 0.5* | 35.6 ± 0.5 * | 32.0 ± 0.5 * | 23.1 ± 0.4 | ||
| 12 h | 0 | 2.3 ± 0.2 | 54.5 ± 0.6 | 21.4 ± 0.3 | 20.1 ± 0.3 | |
| 8f | 10 | 6.5 ± 0.2 * | 47.8 ± 1.0 | 22.4 ± 0.2 | 21.5 ± 1.2 | |
| 30 | 17.8 ± 1.0 * | 27.4 ± 0.4 * | 25.5 ± 0.1 * | 27.6 ± 0.6 * | ||
| 8k | 10 | 12.1 ± 0.8 * | 41.9 ± 0.1 * | 20.2 ± 0.4 | 23.7 ± 1.4 | |
| 30 | 24.3 ± 1.5 * | 17.3 ± 0.4 * | 26.8 ± 0.2 * | 29.9 ± 1.7 * | ||
| 24 h | 0 | 3.4 ± 0.2 | 53.6 ± 0.2 | 24.5 ± 0.5 | 16.2 ± 0.5 | |
| 8f | 10 | 10.3 ± 0.7 * | 42.5 ± 0.3 * | 29.8 ± 0.3 * | 15.0 ± 0.8 | |
| 30 | 56.2 ± 2.0 * | 12.6 ± 0.7 * | 13.6 ± 0.6 * | 16.6 ± 0.8 | ||
| 8k | 10 | 12.7 ± 0.1 * | 39.4 ± 0.2 * | 28.4 ± 1.1 * | 17.2 ± 0.6 | |
| 30 | 51.5 ± 4.2 * | 11.2 ± 0.9 * | 11.5 ± 0.4 * | 23.8 ± 2.9 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, P.; Onnis, V.; Balboni, E.; Hamel, E.; Estévez-Sarmiento, F.; Quintana, J.; Estévez, F.; Brancale, A.; Ferla, S.; Manfredini, S.; et al. Synthesis and Biological Evaluation of 2-Substituted Benzyl-/Phenylethylamino-4-amino-5-aroylthiazoles as Apoptosis-Inducing Anticancer Agents. Molecules 2020, 25, 2177. https://doi.org/10.3390/molecules25092177
Oliva P, Onnis V, Balboni E, Hamel E, Estévez-Sarmiento F, Quintana J, Estévez F, Brancale A, Ferla S, Manfredini S, et al. Synthesis and Biological Evaluation of 2-Substituted Benzyl-/Phenylethylamino-4-amino-5-aroylthiazoles as Apoptosis-Inducing Anticancer Agents. Molecules. 2020; 25(9):2177. https://doi.org/10.3390/molecules25092177
Chicago/Turabian StyleOliva, Paola, Valentina Onnis, Elisa Balboni, Ernest Hamel, Francisco Estévez-Sarmiento, José Quintana, Francisco Estévez, Andrea Brancale, Salvatore Ferla, Stefano Manfredini, and et al. 2020. "Synthesis and Biological Evaluation of 2-Substituted Benzyl-/Phenylethylamino-4-amino-5-aroylthiazoles as Apoptosis-Inducing Anticancer Agents" Molecules 25, no. 9: 2177. https://doi.org/10.3390/molecules25092177