
Structure–Activity Relationship Studies on Novel Antiviral Agents for Norovirus Infections

 , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
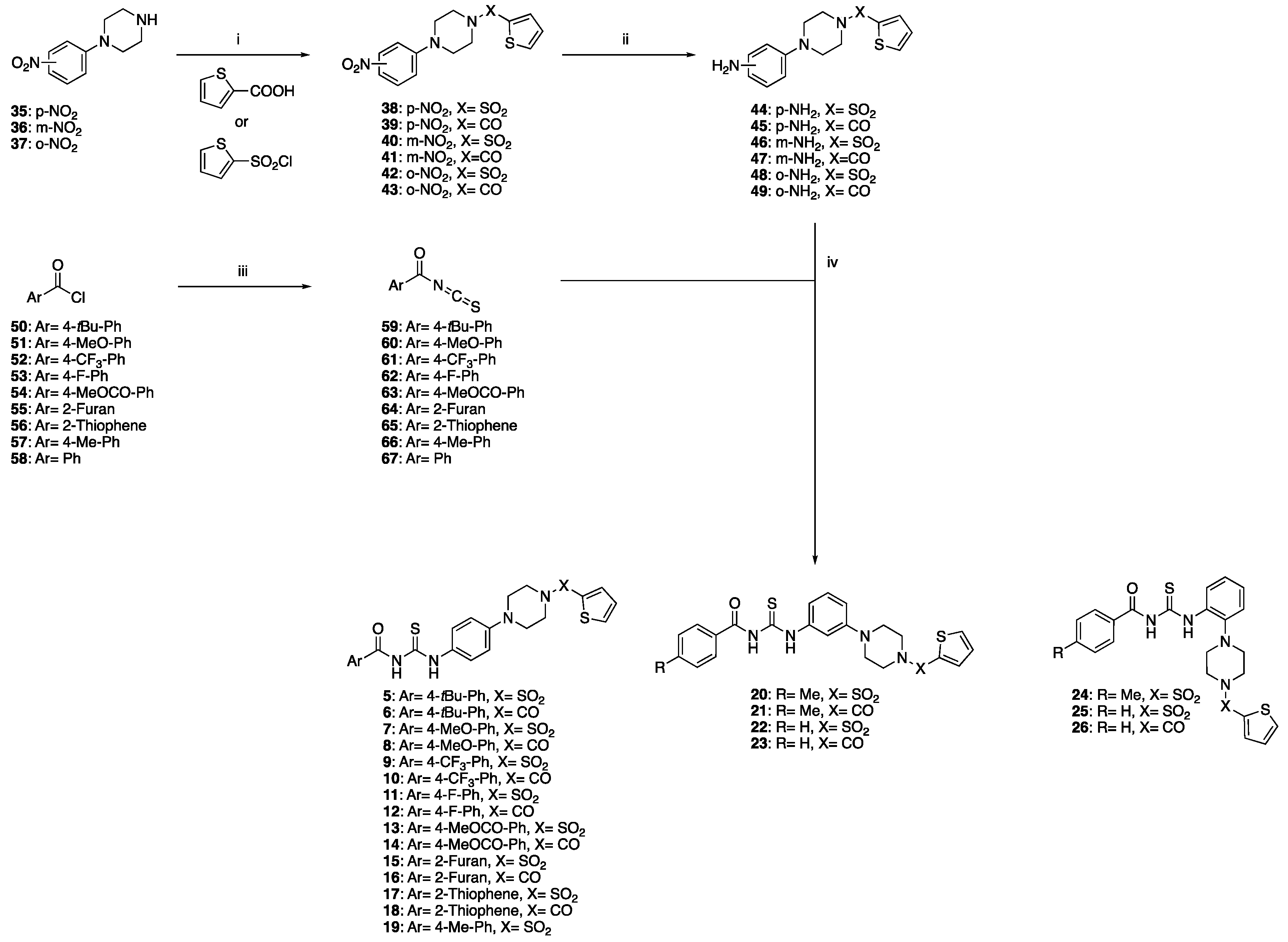
2.1. Synthetic Chemistry
2.1.1. General Procedure for the Preparation of Target Products 5–26
- 4-(tert-Butyl)-N-((4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (5)
- 4-(tert-Butyl)-N-((4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (6)
- 4-Methoxy-N-((4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (7)
- 4-Methoxy-N-((4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (8)
- N-((4-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)-4-(trifluoromethyl)benzamide (9)
- N-((4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)-4-(trifluoromethyl)benzamide (10)
- 4-Fluoro-N-((4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (11)
- 4-Fluoro-N-((4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (12)
- Methyl.4-(((4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)carbamoyl)benzoate (13)
- Methyl 4-(((4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)carbamoyl)benzoate (14)
- N-((4-(4-Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)furan-2-carboxamide (15)
- N-((4-(4-(Thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)furan-2-carboxamide (16)
- N-((4-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)thiophene-2-carboxamide (17)
- N-((4-(4-(Thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)thiophene-2-carboxamide (18)
- 4-Methyl-N-((4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (19)
- 4-Methyl-N-((3-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (20)
- 4-Methyl-N-((3-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (21)
- N-((3-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (22)
- N-((3-(4-(Thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (23)
- 4-Methyl-N-((2-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (24)
- N-((2-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (25)
- N-((2-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (26)
2.1.2. General Procedure for the Preparation of Target Products 27–34
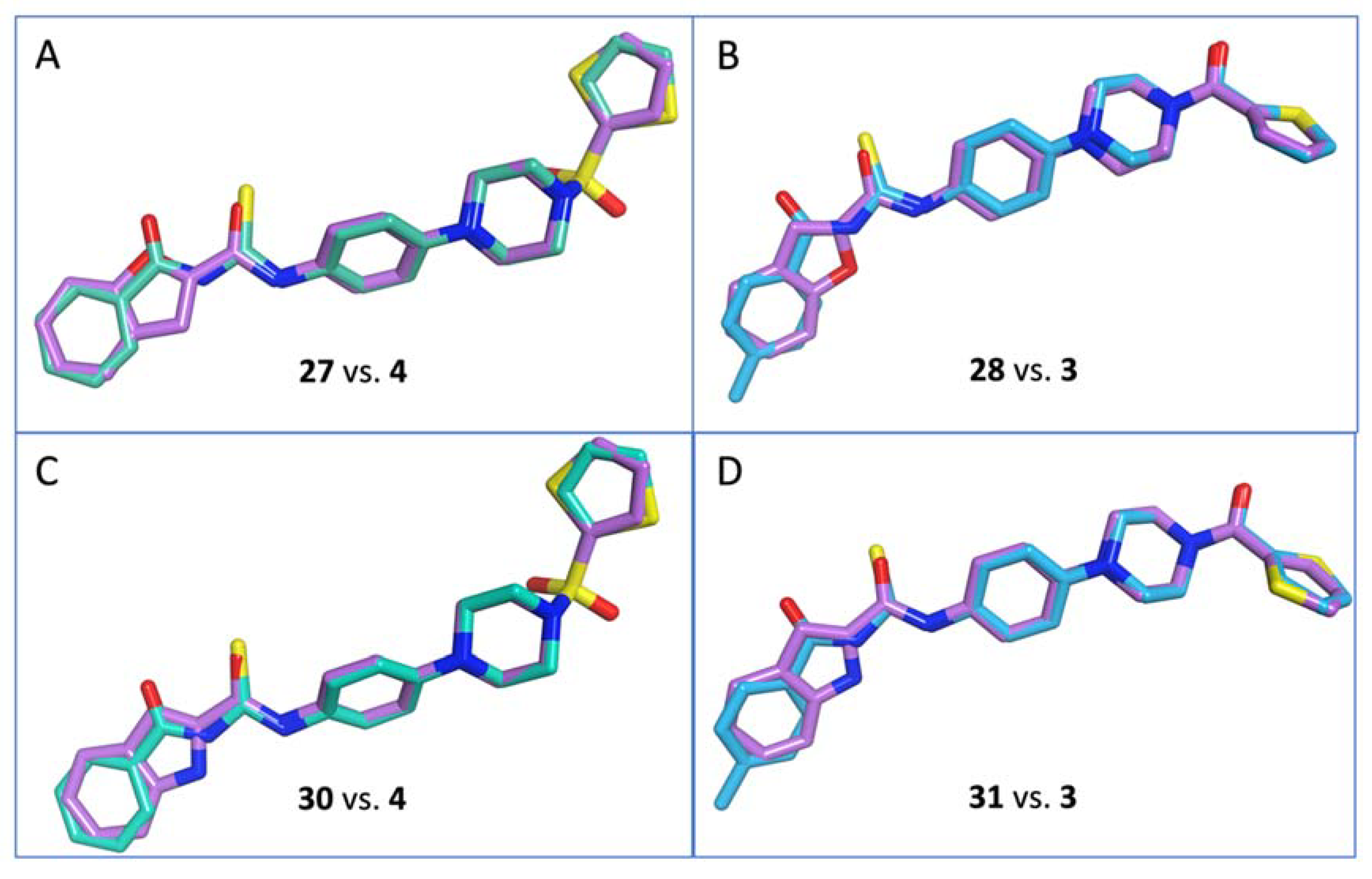
- N-(4-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)benzofuran-2-carboxamide (27)
- N-(4-(4-(Thiophene-2-carbonyl)piperazin-1-yl)phenyl)benzofuran-2-carboxamide (28)
- 5-Methoxy-N-(4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)benzofuran-2-carboxamide (29)
- N-(4-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)-1H-indole-2-carboxamide (30)
- N-(4-(4-(Thiophene-2-carbonyl)piperazin-1-yl)phenyl)-1H-indole-2-carboxamide (31)
- 5-Methyl-N-(4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)-1H-indole-2-carboxamide (32)
- 5-Methoxy-N-(4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)-1H-indole-2-carboxamide (33)
- 6-Methoxy-N-(4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)phenyl)-1H-indole-2-carboxamide (34)
2.2. Antiviral Assays
2.3. Molecular Modelling
3. Results and Discussion
3.1. Design and Synthesis of Novel Antiviral Analogues
3.2. Cell-Based Antiviral Studies against MNV and HuNoV
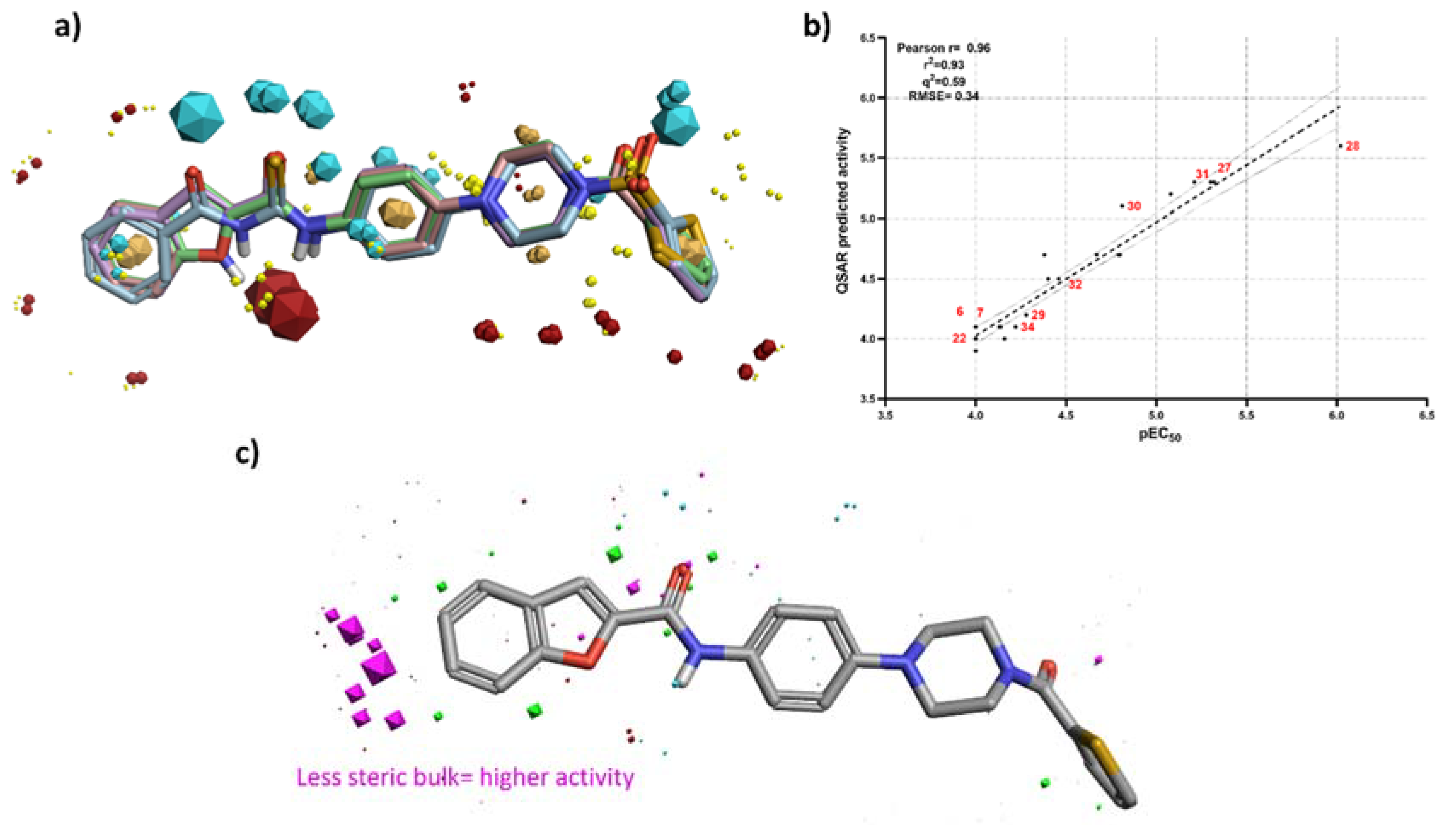
3.3. 3D-QSAR Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robilotti, E.; Deresinski, S.; Pinsky, B.A. Norovirus. Clin. Microbiol. Rev. 2015, 28, 134–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desselberger, U. Viral gastroenteritis. Medicine 2017, 45, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Khamrin, P.; Maneekarn, N. Epidemiology and genotypic distribution of noroviruses in patients with acute gastroenteritis in developing and developed countries. Ann. Res. Hosp. 2019, 3, 1. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global Economic Burden of Norovirus Gastroenteritis. PLoS ONE 2016, 11, 1–16. [Google Scholar]
- Randazzo, W.; D’Souza, D.H.; Sanchez, G. Norovirus: The burden of the unknown. Adv. Food Nutr. Res. 2018, 86, 13–53. [Google Scholar] [PubMed]
- Chhabra, P.; De Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Galasiti Kankanamalage, A.C.; Chang, K.O.; Groutas, W.C. Recent Advances in the Discovery of Norovirus Therapeutics. J. Med. Chem. 2015, 58, 9438–9450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, S.; Principi, N. Norovirus Vaccine: Priorities for Future Research and Development. Front. Immunol. 2020, 11, 1383. [Google Scholar] [CrossRef] [PubMed]
- Netzler, N.E.; Tuipulotu, D.E.; White, P.A. Norovirus antivirals: Where are we now? Med. Res. Rev. 2019, 39, 860–886. [Google Scholar] [CrossRef] [PubMed]
- Chimerix. Chimerix Announces Discovery and Demonstrated Preclinical Activity Supporting Ongoing Phase 1 Study of New Antiviral for Treatment and Prevention of Norovirus. 2018. Available online: http://ir.chimerix.com/news-releases/news-release-details/chimerix-announces-discovery-and-demonstrated-preclinical (accessed on 30 June 2021).
- Lanier, R.S.D.; Kolawole, A.; Hosmillo, M.; Nayak, K.; Bae, A.; Gurley, S.; Tippin, T.; Colton, H.; Dunn, J.; Mullin, M. CMX521: A nucleoside with pan-genotypic activity against norovirus. In Proceedings of the 31st International Conference on Antiviral Research, Porto, Portugal, 11–15 June 2018. [Google Scholar]
- Adamson, C.S.; Chibale, K.; Goss, R.J.M.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral drug discovery: Preparing for the next pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Bassetto, M.; Van Dycke, J.; Neyts, J.; Brancale, A.; Rocha-Pereira, J. Targeting the viral polymerase of diarrhea-causing viruses as a strategy to develop a single broad-spectrum antiviral therapy. Viruses 2019, 11, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferla, S.; Netzler, N.E.; Ferla, S.; Veronese, S.; Tuipulotu, D.E.; Guccione, S.; Brancale, A.; White, P.A.; Bassetto, M. In silico screening for human norovirus antivirals reveals a novel non-nucleoside inhibitor of the viral polymerase. Sci. Rep. 2018, 8, 4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giancotti, G.; Rigo, I.; Pasqualetto, G.; Young, M.T.; Neyts, J.; Rocha-Pereira, J.; Brancale, A.; Ferla, S.; Bassetto, M. A new antiviral scaffold for human norovirus identified with computer-aided approaches on the viral polymerase. Sci. Rep. 2019, 9, 18413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha-Pereira, J.; Jochmans, D.; Debing, Y.; Verbeken, E.; Nasvimento, M.S.J.; Neyts, J. The viral polymerase Inhibitor 2′-C-Methylcytidine inhibits norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J. Virol. 2013, 87, 11798–11805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE 2020.10). Chemical Computing Group, Inc.: Montreal, QC, Canada. Available online: http://www.chemcomp.com (accessed on 16 July 2021).
- Forge, Version 10, Cresset®, Litlington, Cambridgeshire, UK. Available online: http://www.cresset-group.com/forge/ (accessed on 16 July 2021).
- ChemAxon. Available online: https://www.chemaxon.com (accessed on 16 July 2021).
- Kolawole, A.O.; Rocha-Pereira, J.; Elftman, M.D.; Neyts, J.; Wobus, C.E. Inhibition of human norovirus by a viral polymer-ase inhibitor in the B cell culture system and in the mouse model. Antivir. Res. 2016, 132, 46–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, M.; Hosmillo, M.; Emmott, E.; Lu, J.; Goodfellow, I. Selection and characterization of rupintrivir-resistant norwalk virus replicon cells in vitro. Antimicrob. Agents Chemother. 2018, 62, e00201-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha-Pereira, J.; Nascimento, M.S.J.; Ma, Q.; Hilgenfeld, R.; Neyts, J.; Jochmans, D. The enterovirus protease inhibitor rupintrivir exerts cross-genotypic anti-norovirus activity and clears cells from the norovirus replicon. Antimicrob. Agents Chemother. 2014, 58, 4675–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Structure | MNV EC50A (µM) | MNV CC50A (µM) | HuNoV EC50A (µM) | HuNoV CC50A (µM) |
|---|---|---|---|---|---|
 | |||||
| 3 | Ar = 4-Me-Ph, X = CO | 46.6 ± 0.3 | >100 | 15.8 ± 13.5 | 64.5 ± 31.5 |
| 4 | Ar= Ph, X= SO2 | 44.4 ± 9.5 | >100 | 4.8 ± 5.2 | 63.9 ± 22.5 |
| 5 | Ar = 4-tBu-Ph, X = SO2 | >100 | 78.80 ± 27.5 | >100 | >100 |
| 6 | Ar = 4-tBu-Ph, X = CO | >100 | 80.5 ± 33.8 | >100 | >100 |
| 7 | Ar = 4-MeO-Ph, X = SO2 | 81.1 ± 30.1 | >100 | >100 | >100 |
| 8 | Ar = 4-MeO-Ph, X = CO | 26.1 ± 11.3 | >100 | 39.5 ± 52.4 | 8.44 ± 3.73 |
| 9 | Ar = 4-CF3-Ph, X = SO2 | 97.5 ± 4.9 | >100 | 73.5 ± 46.0 | 11.21 ± 6.31 |
| 10 | Ar = 4-CF3-Ph, X = CO | 75.3 ± 29.1 | >100 | >100 | 11.67 ± 9.06 |
| 11 | Ar = 4-F-Ph, X = SO2 | >100 | 59.9 ± 31.4 | 21.4 ± 9.6 | 15.26 ± 11.12 |
| 12 | Ar = 4-F-Ph, X = CO | >100 | >100 | 41.3 ± 50.9 | 29.53 ± 14.16 |
| 13 | Ar = 4-MeOCO-Ph X = SO2 | >100 | >100 | 16.4 ± 13.3 | >100 |
| 14 | Ar = 4-MeOCO-Ph X = CO | 46.6 ± 12.1 | >100 | >100 | >100 |
| 15 | Ar = 2-Furan, X = SO2 | 66.0 ± 35.6 | >100 | 58.7 ± 36.9 | 63.50 ± 31.81 |
| 16 | Ar = 2-Furan, X = CO | >100 | >100 | >100 | 67.71 ± 24.49 |
| 17 | Ar = 2-Thiophene X = SO2 | 34.7 ± 18.1 | >100 | 45.4 ± 47.3 | 17.82 ± 5.00 |
| 18 | Ar = 2-Thiophene, X = CO | 60.5 ± 29.2 | >100 | >100 | >100 |
| 19 | Ar = 4-Me-Ph, X = SO2 | >100 | 49.8 ± 35.6 | 71.7 ± 49.1 | 37.42 ± 11.84 |
 | |||||
| 20 | R = Me, X = SO2 | 78.2 ± 27.3 | >100 | >100 | >100 |
| 21 | R = Me, X = CO | 92.7 ± 9.2 | >100 | >100 | 81.3 ± 37.5 |
| 22 | R= H, X = SO2 | 99.0 ± 1.7 | >100 | >100 | >100 |
| 23 | R = H, X = CO | >100 | >100 | >100 | 68.8 ± 37.5 |
 | |||||
| 24 | R = Me, X = SO2 | 34.0 ± 30.1 | >100 | 80.4 ± 23.5 | >100 |
| 25 | R = H, X = SO2 | 77.5 ± 15.4 | >100 | 17.4 ± 16.2 | >100 |
| 26 | R = H, X = CO | 72.9 ± 46.9 | >100 | 9.4 ± 4.2 | 87.5 ± 25.0 |
 | |||||
| 27 | R = H, Y = O, X = SO2 | 64.3 ± 41.3 | >100 | 5.0 ± 1.9 | 22.0 ± 7.4 |
| 28 | R = H, Y = O, X = CO | 70.8 ± 30.2 | >100 | 0.9 ± 0.4 | 4.1 ± 1.7 |
| 29 | R = 5-OMe, Y = O, X = CO | >100 | 93.2 ± 11.8 | 52.5 ± 32.1 | 87.5 ± 25.0 |
| 30 | R = H, Y = NH, X = SO2 | 23.7 ± 12.4 | >100 | 15.6 ± 3.3 | 70.0 ± 21.7 |
| 31 | R = H, Y = NH, X = CO | 25.9 ± 14.2 | >100 | 6.2 ± 4.4 | 18.2 ± 9.3 |
| 32 | R = 5-Me, Y = NH, X = SO2 | 25.8 ± 14.6 | >100 | 34.9 ± 6.1 | >100 |
| 33 | R = 5-OMe, Y = NH X = SO2 | 25.5 ± 14.3 | >100 | 68.9 ± 37.0 | >100 |
| 34 | R = 6-OMe, Y = NH X = SO2 | 26.2 ± 14.7 | >100 | 60.4 ± 30.2 | >100 |
| 2CMC | Positive control | 16.2 ± 5.9 | 36.5 ± 7.7 | n.d.B | n.d.B |
| Rupintrivir | Positive control | n.d.B | n.d.B | 1.5 ± 0.2 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferla, S.; Varricchio, C.; Knight, W.; Ho, P.K.; Saporito, F.; Tropea, B.; Fagan, G.; Flude, B.M.; Bevilacqua, F.; Santos-Ferreira, N.; et al. Structure–Activity Relationship Studies on Novel Antiviral Agents for Norovirus Infections. Microorganisms 2021, 9, 1795. https://doi.org/10.3390/microorganisms9091795
Ferla S, Varricchio C, Knight W, Ho PK, Saporito F, Tropea B, Fagan G, Flude BM, Bevilacqua F, Santos-Ferreira N, et al. Structure–Activity Relationship Studies on Novel Antiviral Agents for Norovirus Infections. Microorganisms. 2021; 9(9):1795. https://doi.org/10.3390/microorganisms9091795
Chicago/Turabian StyleFerla, Salvatore, Carmine Varricchio, William Knight, Pui Kei Ho, Fabiana Saporito, Beatrice Tropea, Giulio Fagan, Ben Matthew Flude, Federica Bevilacqua, Nanci Santos-Ferreira, and et al. 2021. "Structure–Activity Relationship Studies on Novel Antiviral Agents for Norovirus Infections" Microorganisms 9, no. 9: 1795. https://doi.org/10.3390/microorganisms9091795