
SARS-CoV-2 Virus−Host Interaction: Currently Available Structures and Implications of Variant Emergence on Infectivity and Immune Response

 , , , and
, , , and
Abstract
:1. Introduction
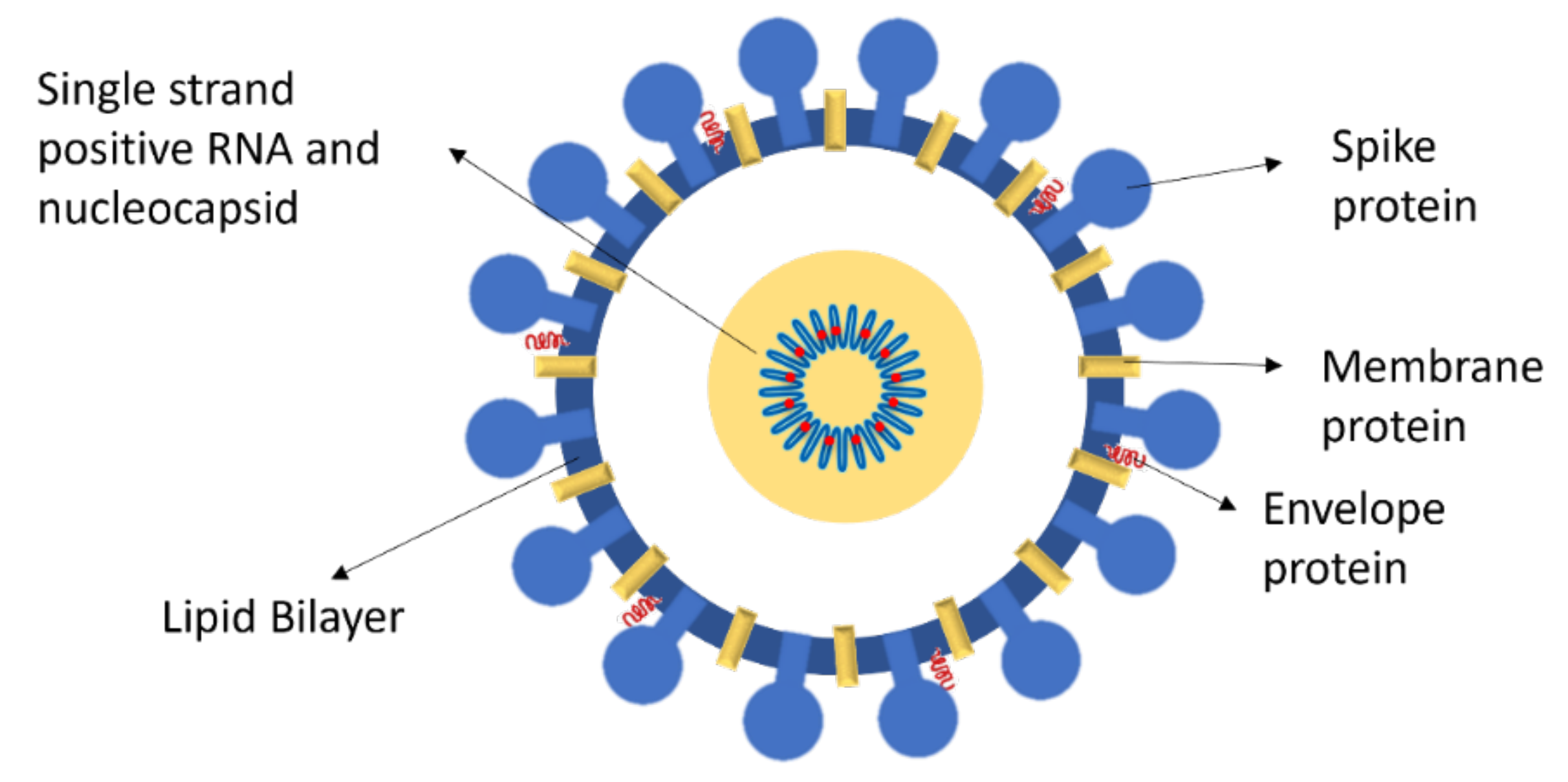
Glycoprotein: Cell Recognition and Infection
2. Discussion
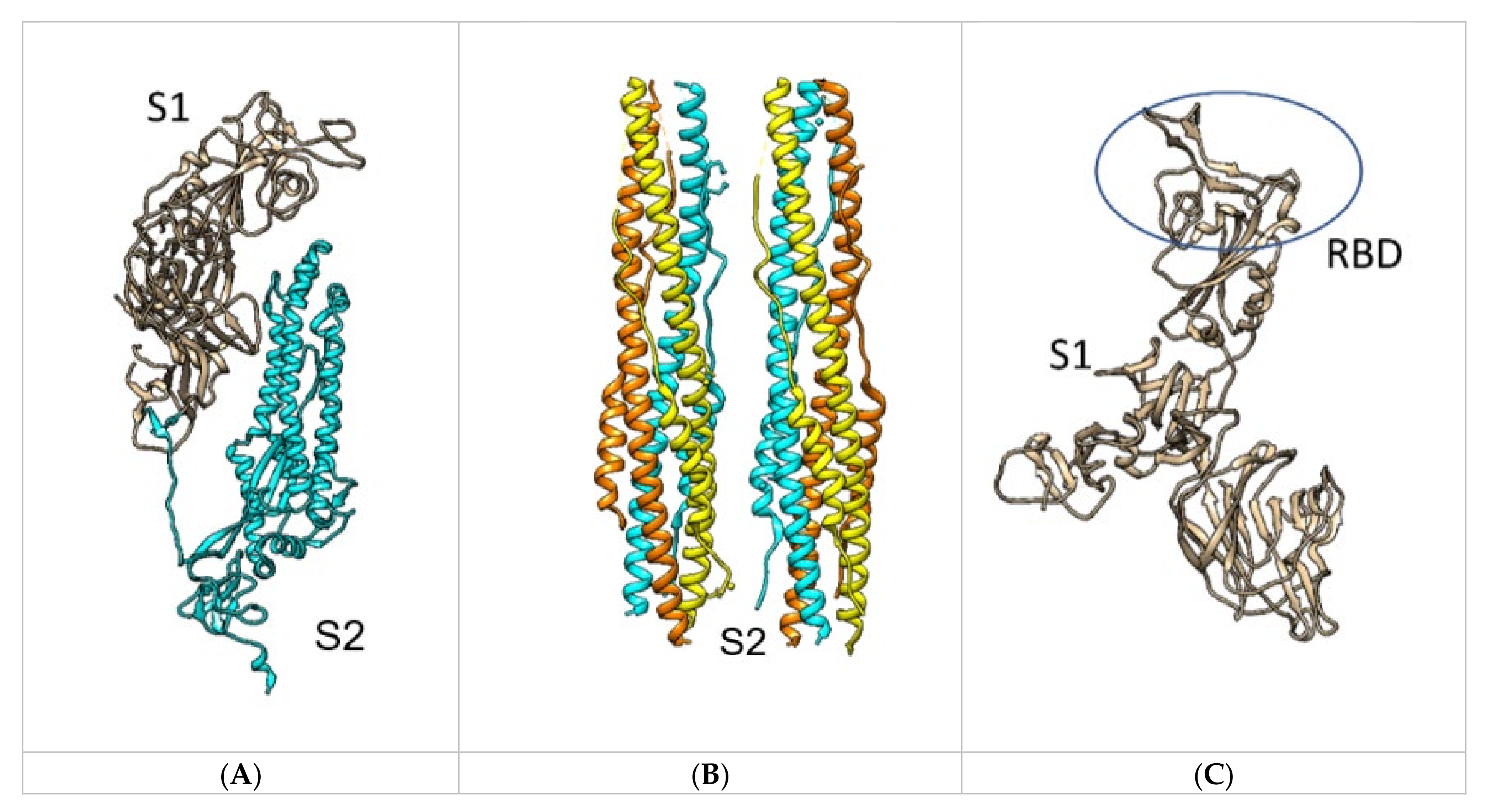
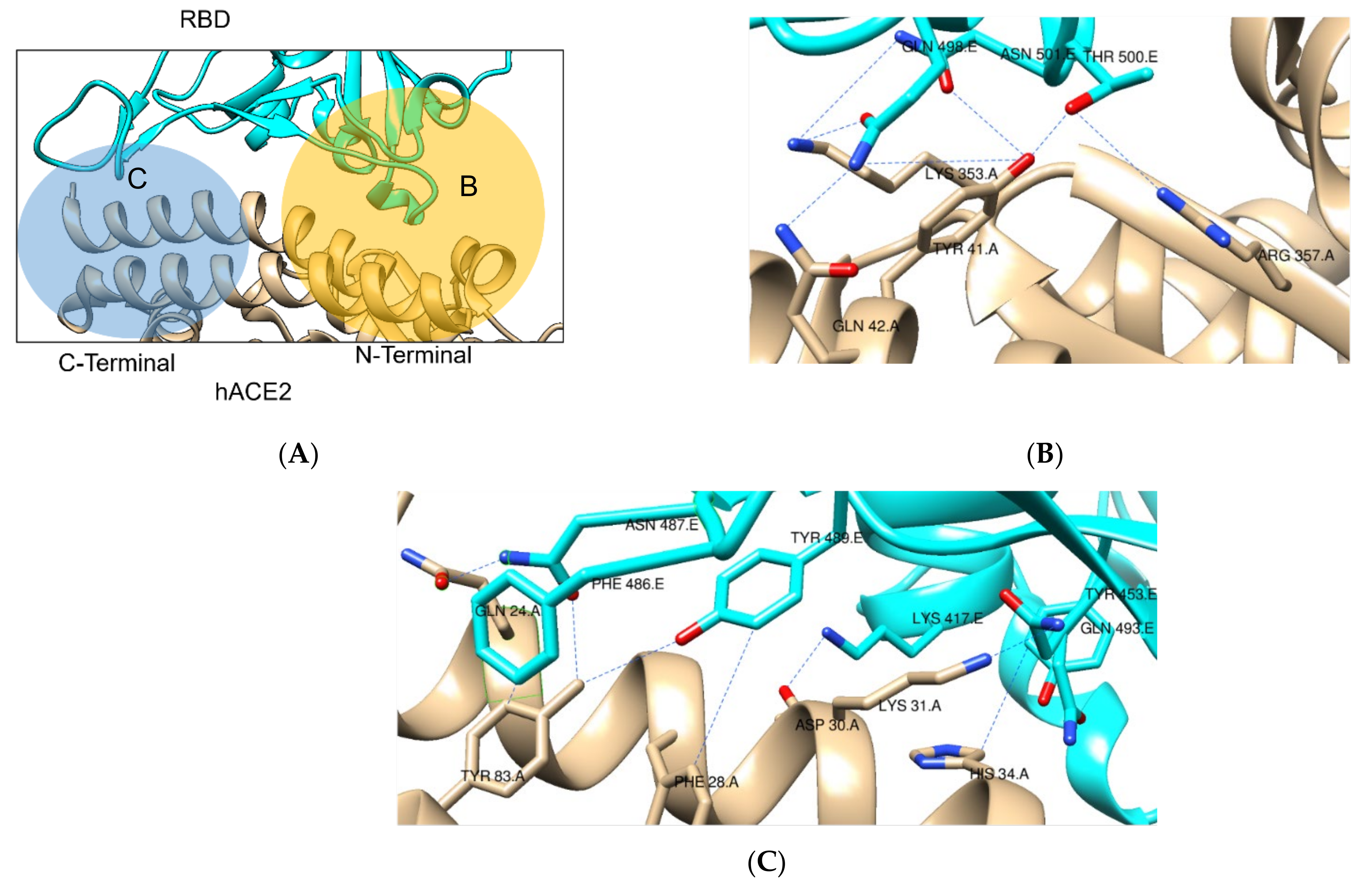
2.1. SARS-CoV-2 Spike RBD – hACE2 Interaction
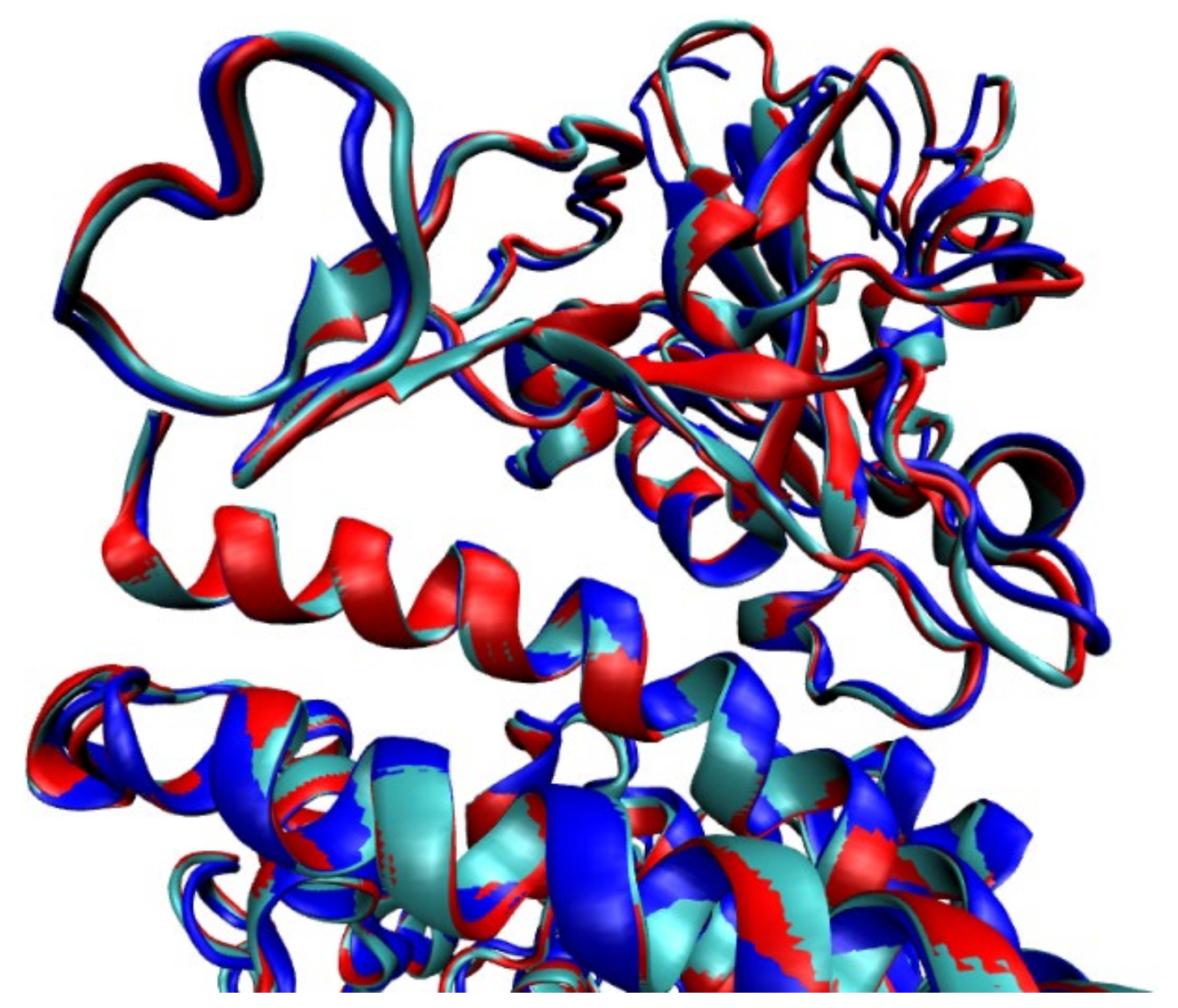
2.1.1. Structures with No Mutation
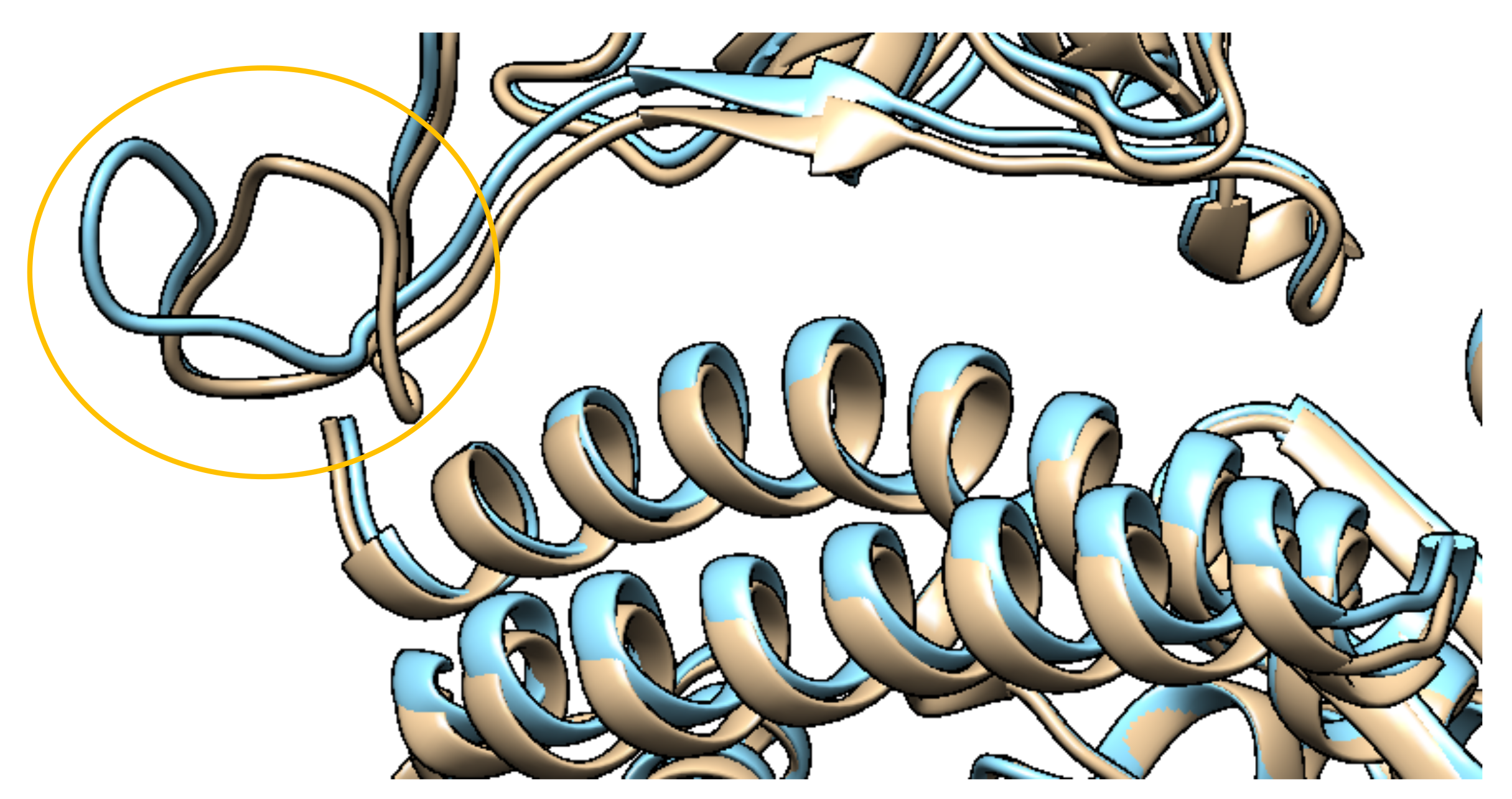
2.1.2. Structures with Mutations
2.1.3. RBD Only Structures—Final Remarks
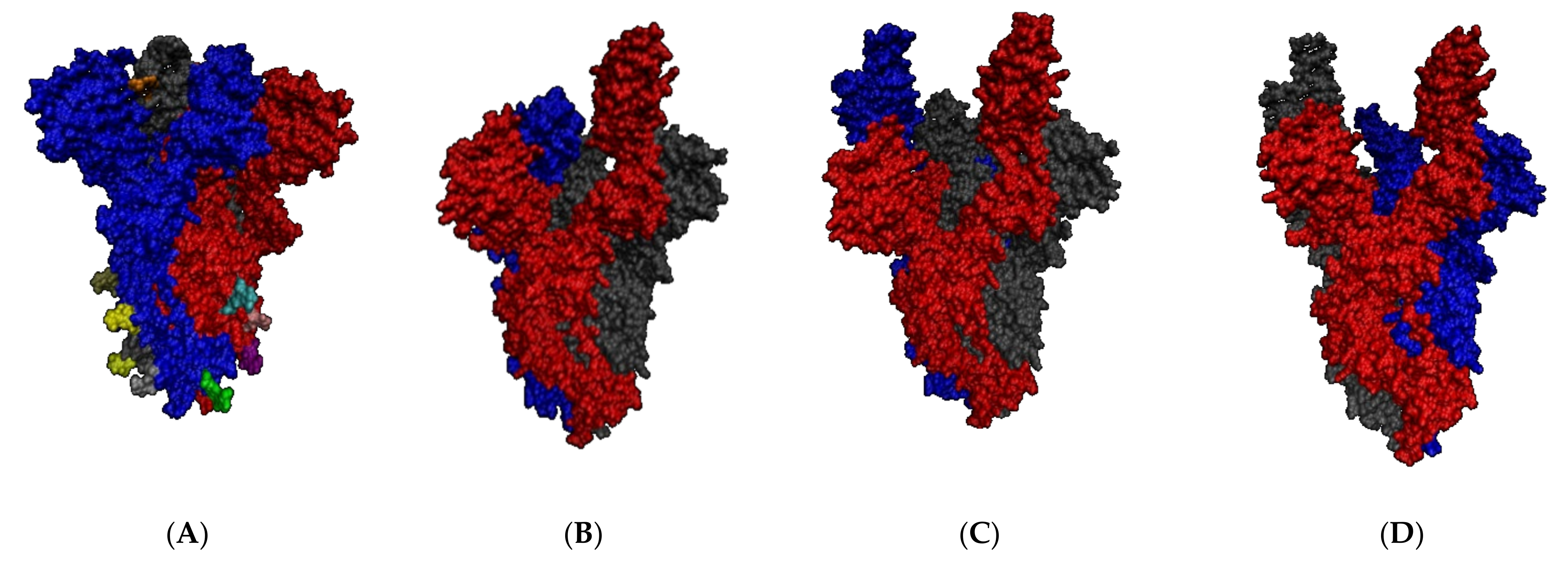
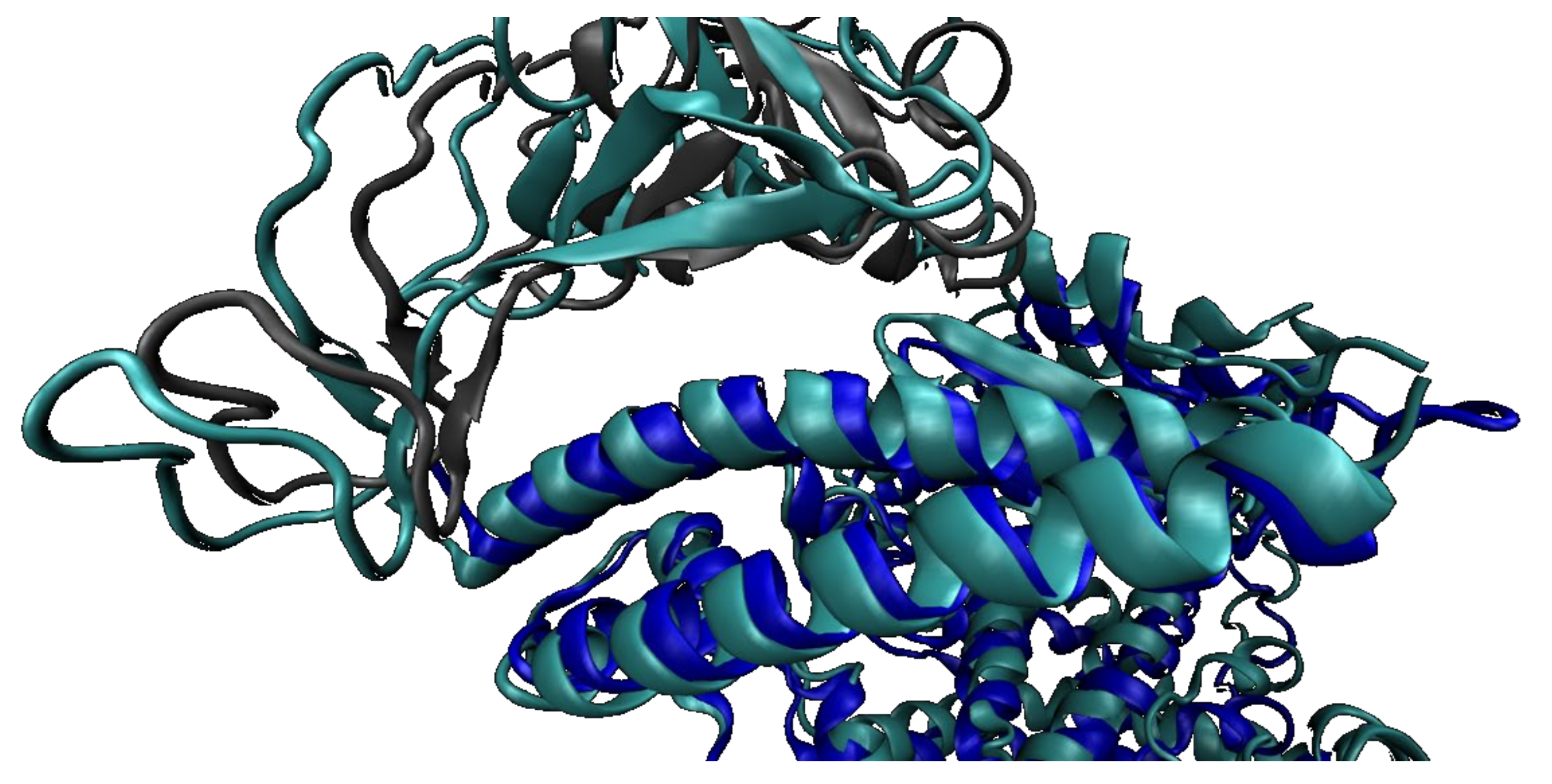
2.2. SARS-CoV-2 Full Spike Protein—hACE2 Interaction
2.2.1. Mutations in the Spike Protein
2.2.2. SARS-CoV-2 Full Spike Protein—hACE2
2.2.3. Before and after hACE2 Binding
2.2.4. Spike Protein Mutation D614
2.2.5. Full Spike Protein Neutralization Strategies
2.2.6. Spike Vaccine Candidate
2.3. Delta Variant
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front Cell Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19-11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 3 September 2021).
- WHO. Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 4 September 2021).
- Zhao, J.; Cui, W.; Tian, B.-p. The Potential Intermediate Hosts for SARS-CoV-2. Front. Microbiol. 2020, 11, 2400. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeman, D.; Gordon, B.; Fielding, B.C. Pathogenic Human Coronaviruses. Ref. Modul. Biomed. Sci. 2021, B978-970-912-818731-818739.800052-818735. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-f.; Xu, W.; Liu, S.-w. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Fan, Y.; Zhao, K.; Shi, Z.-L.; Zhou, P. Bat Coronaviruses in China. Viruses 2019, 11, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Coronavirus disease (COVID-19): How is it transmitted? Available online: https://www.who.int/news-room/q-a-detail/coronavirus-disease-covid-19-how-is-it-transmitted (accessed on 18 August 2021).
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens 2021, 10, 633. [Google Scholar] [CrossRef]
- ECDC. SARS-CoV-2 Variants of Concern as of 9 September 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 15 September 2021).
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 14 September 2021).
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html#Concern (accessed on 14 September 2021).
- CDC. Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/ (accessed on 29 July 2021).
- ECDC. European Centre for Disease Prevention and Control. Available online: https://www.ecdc.europa.eu/en (accessed on 29 July 2021).
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. New Engl. J. Med. 2021, 384, 1885–1898. [Google Scholar] [CrossRef]
- Cele, S.; Gazy, I.; Jackson, L.; Hwa, S.-H.; Tegally, H.; Lustig, G.; Giandhari, J.; Pillay, S.; Wilkinson, E.; Naidoo, Y.; et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 2021, 593, 142–146. [Google Scholar] [CrossRef]
- Pearson, C.A.; Russell, T.W.; Davies, N.G. Estimates of Severity and Transmissibility of Novel South Africa SARS-CoV-2 Varian 501Y.V2. Available online: https://cmmid.github.io/topics/covid19/reports/sa-novel-variant/2021_01_11_Transmissibility_and_severity_of_501Y_V2_in_SA.pdf (accessed on 14 September 2021).
- Funk, T.; Pharris, A.; Spiteri, G.; Bundle, N.; Melidou, A.; Carr, M.; Gonzalez, G.; Garcia-Leon, A.; Crispie, F.; O’Connor, L.; et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: Data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Eurosurveillance 2021, 26, 2100348. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef]
- Eli-Lilly. FACT SHEET FOR HEALTH CARE PROVIDERS EMERGENCY USE AUTHORIZATION (EUA) OF BAMLANIVIMAB AND ETESEVIMAB. Available online: https://www.fda.gov/media/145802/download (accessed on 15 September 2021).
- Uriu, K.; Kimura, I.; Shirakawa, K.; Takaori-Kondo, A.; Nakada, T.-a.; Kaneda, A.; Consortium, T.G.t.P.J.; Nakagawa, S.; Sato, K. Ineffective neutralization of the SARS-CoV-2 Mu variant by convalescent and vaccine sera. bioRxiv 2021, 2021.2009.2006.459005. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallum, M.; Marco, A.; Lempp, F.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Huo, J.; Zhao, Y.; Ren, J.; Zhou, D.; Duyvesteyn, H.M.E.; Ginn, H.M.; Carrique, L.; Malinauskas, T.; Ruza, R.R.; Shah, P.N.M.; et al. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe 2020, 28, 445–454.e6. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Delta variant: What is happening with transmission, hospital admissions, and restrictions? BMJ 2021, 373, n1513. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Chiaravalli, J.; Meyer, B.; Dym, O.; Elad, N.; Schreiber, G. SARS-CoV-2 RBD in vitroevolution follows contagious mutation spread, yet generates an able infection inhibitor. bioRxiv 2021, 2021.2001.2006.425392. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Delgado Blanco, J.; Hernandez-Alias, X.; Cianferoni, D.; Serrano, L. In silico mutagenesis of human ACE2 with S protein and translational efficiency explain SARS-CoV-2 infectivity in different species. PLoS Comput. Biol. 2020, 16, e1008450. [Google Scholar] [CrossRef]
- Higuchi, Y.; Suzuki, T.; Arimori, T.; Ikemura, N.; Mihara, E.; Kirita, Y.; Ohgitani, E.; Mazda, O.; Motooka, D.; Nakamura, S.; et al. High affinity modified ACE2 receptors protect from SARS-CoV-2 infection in hamsters. biorXiv 2020. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879.e5. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Bi, W.; Wang, X.; Xu, W.; Yan, R.; Zhang, Y.; Zhao, K.; Li, Y.; Zhang, M.; Cai, X.; et al. Engineered trimeric ACE2 binds viral spike protein and locks it in “Three-up” conformation to potently inhibit SARS-CoV-2 infection. Cell Res. 2021, 31, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; Purcell, D.F.J.; Parker, M.W. SARS-CoV-2 Spike receptor-binding domain with a G485R mutation in complex with human ACE2. bioRxiv 2021, 2021.2003.2016.434488. [Google Scholar] [CrossRef]
- Hodcroft, E. Variant: 20I (Alpha, V1). Available online: https://covariants.org/variants/20I.Alpha.V1 (accessed on 7 July 2021).
- Hodcroft, E. Variant: 20H (Beta, V2). Available online: https://covariants.org/variants/20H.Beta.V2 (accessed on 7 July 2021).
- Hodcroft, E. Variant: 20J (Gamma, V3). Available online: https://covariants.org/variants/20J.Gamma.V3 (accessed on 7 July 2021).
- Juraszek, J.; Rutten, L.; Blokland, S.; Bouchier, P.; Voorzaat, R.; Ritschel, T.; Bakkers, M.J.G.; Renault, L.L.R.; Langedijk, J.P.M. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 2021, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Ye, F.; Guo, Y.; Xia, L.; Zhong, X.; Chi, X.; Zhou, Q. Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 2021, 31, 717–719. [Google Scholar] [CrossRef] [PubMed]
- WHO. SARS-CoV-2 Variants. Available online: https://www.who.int/csr/don/31-december-2020-sars-cov2-variants/en/ (accessed on 3 September 2021).
- Weissman, D.; Alameh, M.G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe. 2021, 29, 23–31.e24. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Lu, J.; Zhang, J.; Johnson, R.I.; McKay, L.G.A.; Storm, N.; Lavine, C.L.; Peng, H.; Cai, Y.; Rits-Volloch, S.; et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef]
- Hodcroft, E. Variant: 21A (Delta). Available online: https://covariants.org/variants/21A.Delta (accessed on 7 July 2021).
- ECDC. SARS-COV-2 Delta variant now dominant in much of the European Region and efforts must be reinforced to prevent transmission, warn WHO/Europe and ECDC. Available online: https://www.ecdc.europa.eu/en/news-events/sars-cov-2-delta-variant-now-dominant-european-region (accessed on 18 August 2021).
- Li, B.; Deng, A.; Li, K.; Hu, Y.; Li, Z.; Xiong, Q.; Liu, Z.; Guo, Q.; Zou, L.; Zhang, H.; et al. Viral infection and transmission in a large well-traced outbreak caused by the Delta SARS-CoV-2 variant. medRxiv 2021, 2021.2007.2007.21260122. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, eabi7994. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen, L.; Dang, H.V.; deMarco, A.; Franko, N.; Tilles, S.W.; Logue, J.; et al. Molecular basis of immune evasion by the delta and kappa SARS-CoV-2 variants. bioRxiv 2021, 2021.2008.2011.455956. [Google Scholar] [CrossRef]
- Hodcroft, E. Variant: 21C (Epsilon). Available online: https://covariants.org/variants/21C.Epsilon (accessed on 8 July 2021).
- Sensoy, O.; Almeida, J.G.; Shabbir, J.; Moreira, I.S.; Morra, G. Chapter 16-Computational studies of G protein-coupled receptor complexes: Structure and dynamics. In Methods in Cell Biology; Shukla, A.K., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 142, pp. 205–245. [Google Scholar]
- NCBI-Virus. SARS-CoV-2 Data Hub. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?VirusLineage_ss=Severe%20acute%20respiratory%20syndrome%20coronavirus%202%20(SARS-CoV-2),%20taxid:2697049&SeqType_s=Nucleotide&Lineage_s=B.1.1.7 (accessed on 31 July 2021).
- Petra, M.; Steven, K.; Mahesh Shanker, D.; Guido, P.; Bo, M.; Swapnil, M.; Charlie, W.; Thomas, M.; Isabella, F.; Rawlings, D.; et al. SARS-CoV-2 B.1.617.2 Delta variant emergence and vaccine breakthrough. Nat. Portf. 2021. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
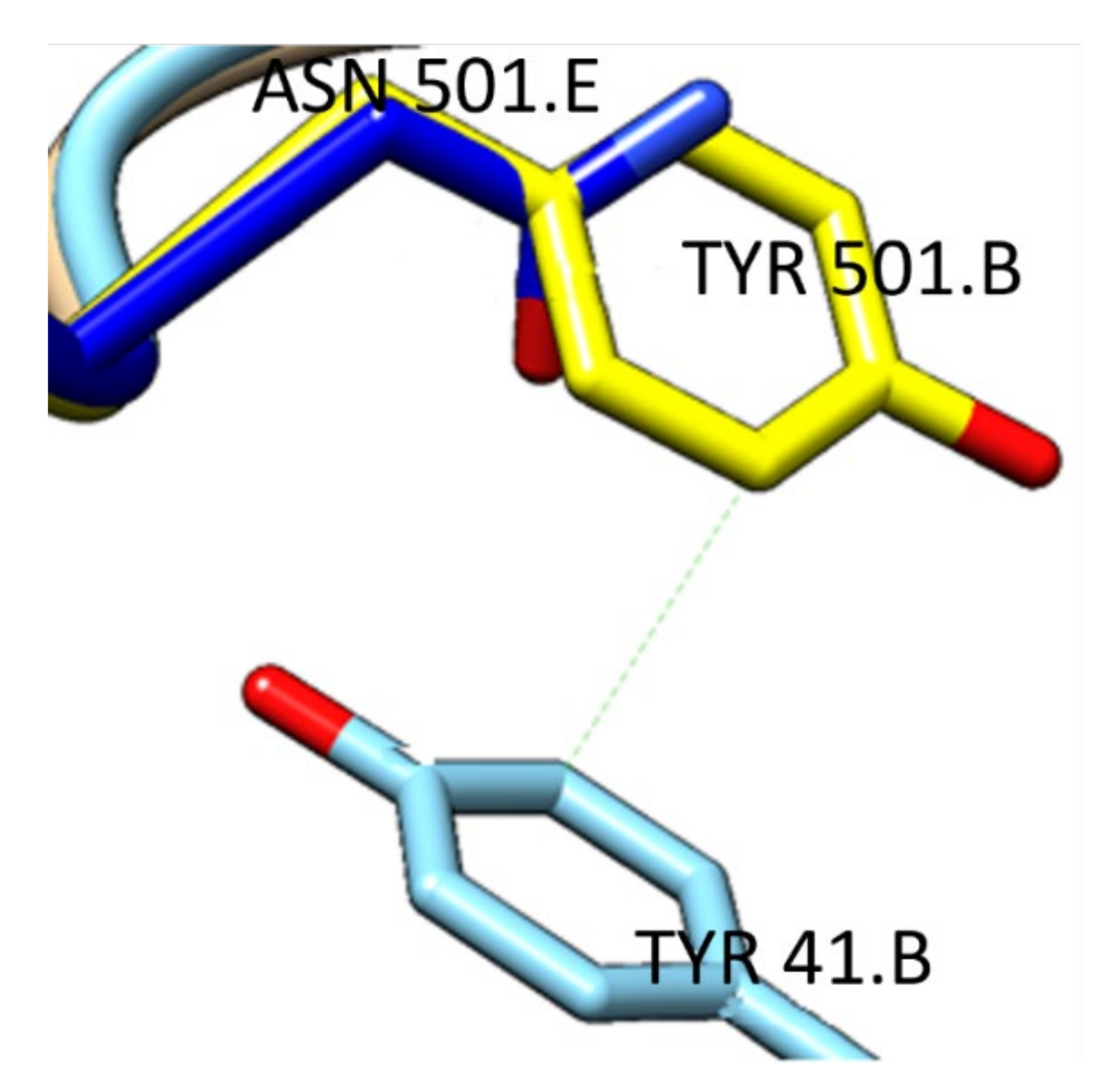{kind=link}
{kind=link}
| WHO Label | Pango Lineage | Origin | Infectivity | Effects on Immune Response | Clinical Relevance | Current Classification (CDC/WHO/ECDC) | References |
|---|---|---|---|---|---|---|---|
| Alpha | B.1.1.7 | United Kingdom Sep-2020 | 50% increased transmission | Minimal impact | Increased severity | VOC | VOC | DE | [14,16] |
| Beta | B.1.351 | South Africa May-2020 | 50% increased transmission | Significant reduction in neutralization | Reduced susceptibility to treatment | VOC | VOC | VOC | [19,20,21,22] |
| Gamma | P.1 | Japan/Brazil Nov-2020 | Potential increased transmissibility1 | Significant reduced neutralization | Significantly reduced susceptibility to treatment | VOC | VOC | VOC | [22,23,24,25] |
| Delta | B.1.617.2 | India Oct-2020 | Increased transmissibility | Significant reduction in neutralization | Reduced susceptibility to treatment | VOC | VOC | VOC | [14,16] |
| Epsilon | B.1.427/B.1.429 | USA Sep-2020 | Unclear | Reduced neutralization | No effect reported | NA | NA | DE | [14] |
| Zeta | P.2 | Brazil Nov-2020 | No effect reported | Potential reduced neutralization1 | No effect reported | NA | NA | DE | [14] |
| Eta | B.1.525 | Nigeria Dec-2020 | No effect reported | Potential reduced neutralization1 | Potential reduced susceptibility to treatment1 | VOI | VOI | DE | [14] |
| Theta | P.3 | Philippines Jan-2021 | Potential increased transmissibility1 | Potential reduced neutralization1 | No effect reported | NA | NA | DE | [14] |
| Iota | B.1.526 | USA Dec-2020 | No effect reported | Reduced neutralization | Potential reduced susceptibility to treatment1 | VOI | VOI | DE | [14,16] |
| Kappa | B.1.617.1 | India Oct-2020 | Potential increased transmissibility1 | Potential reduced neutralization1 | Potential reduced susceptibility to treatment1 | VOI | VOI | DE | [14,16] |
| Lambda | C.37 | Peru Dec-2020 | Potential increased transmissibility1 | Significantly reduction in neutralization | No effect reported | NA | VOI | VOI | [14,26] |
| Mu | B.1.621 | Colombia Jan-2021 | Potential increased transmissibility1 | Reduced neutralization | No effect reported | NA | VOI | VOI | [14] |
| Mutation | Location | Effect on Affinity | Effect on Immune Response | Additional Information | References |
|---|---|---|---|---|---|
| T19R | S1/NTD | No effect reported | Affects NTD- directed Abs | Change in NTD antigenic site | [32,64] |
| E156- | S1/NTD | No effect reported | Affects NTD- directed Abs | ||
| F157- | S1/NTD | No effect reported | Affects NTD- directed Abs | ||
| R158G | S1/NTD | No effect reported | Affects NTD-directed Abs | ||
| K417N | RBD | Potential decrease in affinity | Moderately decreases RBD-directed Abs | Affects only less important class of Abs | [24,52] |
| L452R | RBD | No effect reported | Severely affects RBD-directed Abs | None | [52,64] |
| T478K | RBD | Increases electrostatic complementarity | Moderately decreases RBD-directed Abs | Three additional H-bonds formed | [65,66,67] |
| D614G | S1-S2 | No effect reported | Increases susceptibility to neutralization by RBD-directed Abs | Changes protein dynamics Increased viral loads and transmission | [52,58] |
| P681R | S2 | No effect reported | No effect reported | Increased S1/S2 cleavage and viral fusion | [66] |
| D950N | S2 | No effect reported | No effect reported | Change protein dynamics | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queirós-Reis, L.; Gomes da Silva, P.; Gonçalves, J.; Brancale, A.; Bassetto, M.; Mesquita, J.R. SARS-CoV-2 Virus−Host Interaction: Currently Available Structures and Implications of Variant Emergence on Infectivity and Immune Response. Int. J. Mol. Sci. 2021, 22, 10836. https://doi.org/10.3390/ijms221910836
Queirós-Reis L, Gomes da Silva P, Gonçalves J, Brancale A, Bassetto M, Mesquita JR. SARS-CoV-2 Virus−Host Interaction: Currently Available Structures and Implications of Variant Emergence on Infectivity and Immune Response. International Journal of Molecular Sciences. 2021; 22(19):10836. https://doi.org/10.3390/ijms221910836
Chicago/Turabian StyleQueirós-Reis, Luís, Priscilla Gomes da Silva, José Gonçalves, Andrea Brancale, Marcella Bassetto, and João R. Mesquita. 2021. "SARS-CoV-2 Virus−Host Interaction: Currently Available Structures and Implications of Variant Emergence on Infectivity and Immune Response" International Journal of Molecular Sciences 22, no. 19: 10836. https://doi.org/10.3390/ijms221910836