


Viruses 2024, 16(5), 726; https://doi.org/10.3390/v16050726 - 03 May 2024
Abstract
In 2022, an unprecedented outbreak of mpox raged in several nations. Sequences from the 2022 outbreak reveal a higher nucleotide substitution if compared with the estimated rate for orthopoxviruses. Recently, intra-lesion SNVs (single nucleotide variants) have been described, and these have been suggested
[...] Read more.
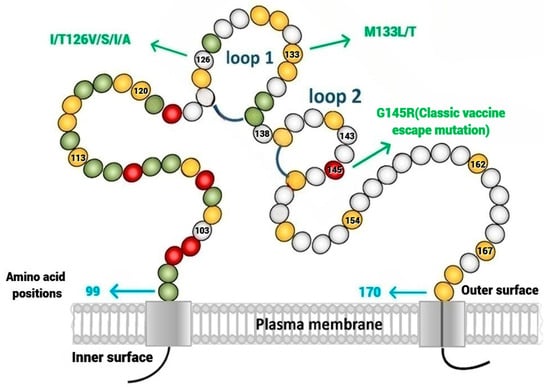
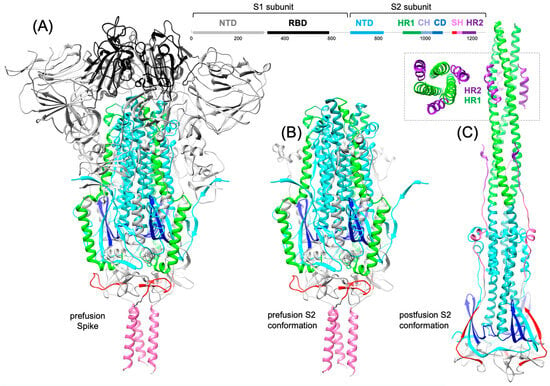
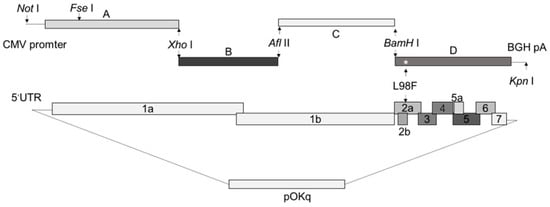
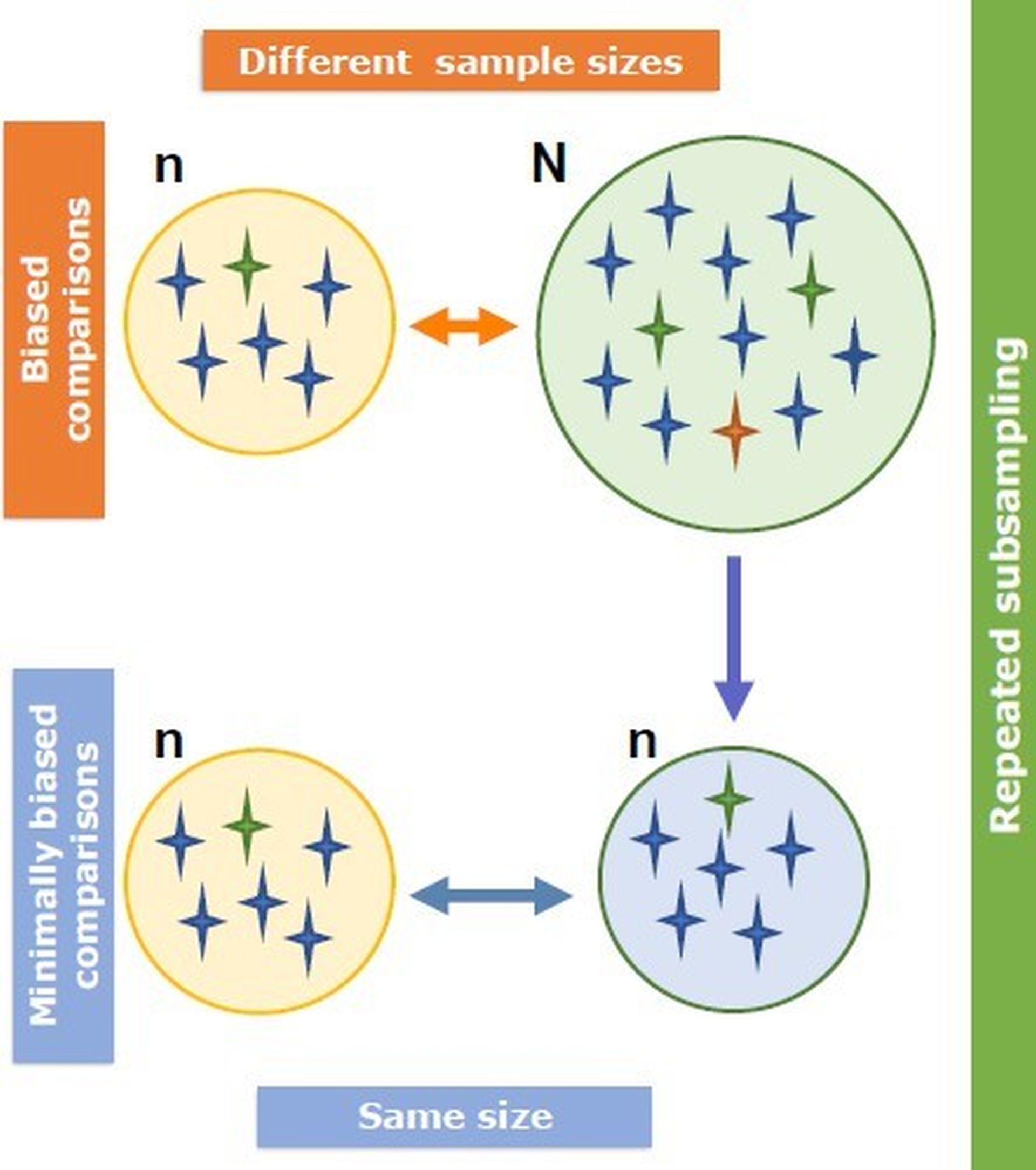
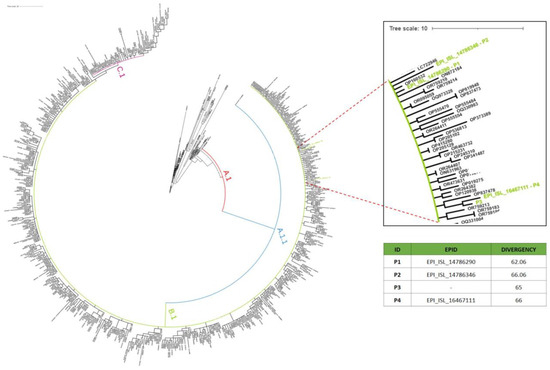
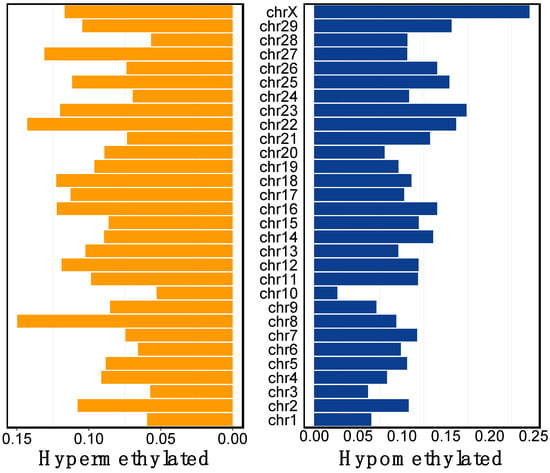
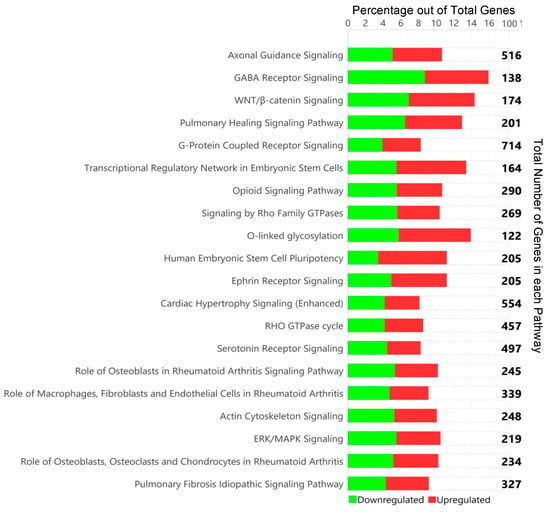
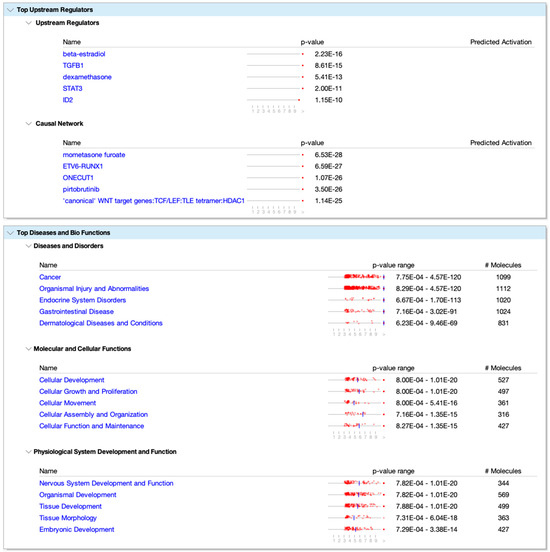
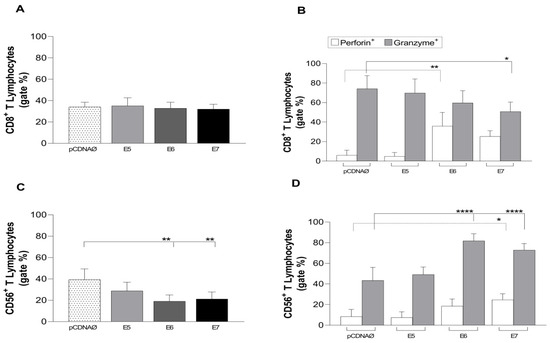
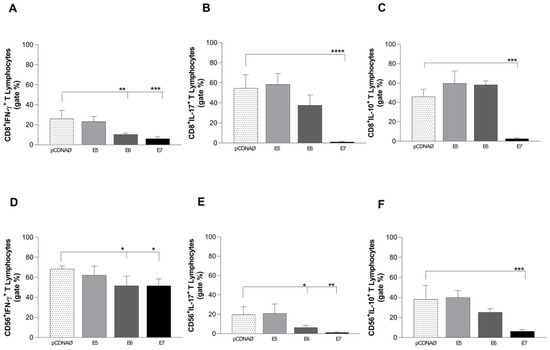
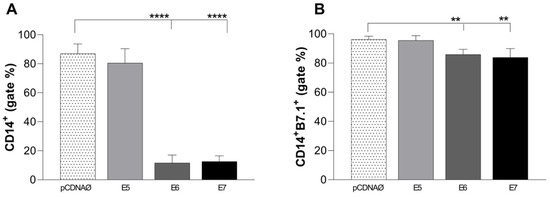
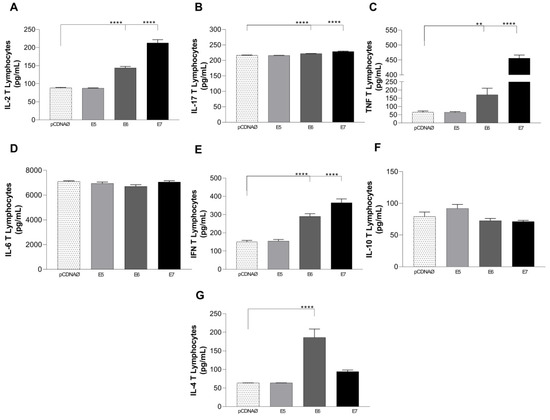
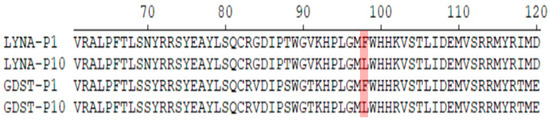
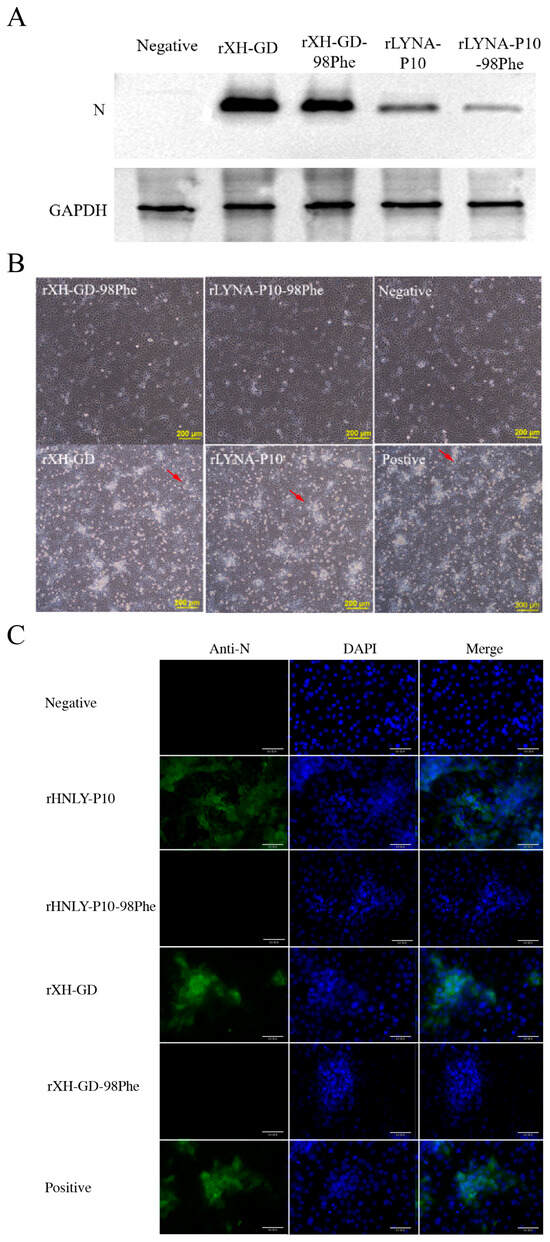
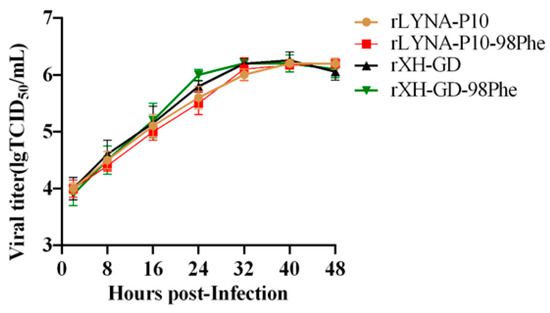
In 2022, an unprecedented outbreak of mpox raged in several nations. Sequences from the 2022 outbreak reveal a higher nucleotide substitution if compared with the estimated rate for orthopoxviruses. Recently, intra-lesion SNVs (single nucleotide variants) have been described, and these have been suggested as possible sources of genetic variation. Until now, it has not been clear if the presence of several SNVs could represents the result of local mutagenesis or a possible co-infection. We investigated the significance of SNVs through whole-genome sequencing analysis of four unrelated mpox cases. In addition to the known mutations harboured by the circulating strains of virus (MPXV), 7 novel mutations were identified, including SNVs located in genes that are involved in immune evasion mechanisms and/or viral fitness, six of these appeared to be APOBEC3-driven. Interestingly, three patients exhibited the coexistence of mutated and wild-type alleles for five non-synonymous variants. In addition, two patients, apparently unrelated, showed an analogous pattern for two novel mutations, albeit with divergent frequencies. The coexistence of mixed viral populations, harbouring non-synonymous mutations in patients, supports the hypothesis of possible co-infection. Additional investigations of larger clinical cohorts are essential to validating intra-patient viral genome heterogeneity and determining the possibility of co-presence events of slightly divergent MPXV strains.
Full article
(This article belongs to the Topic Human Monkeypox Research)
►
Show Figures
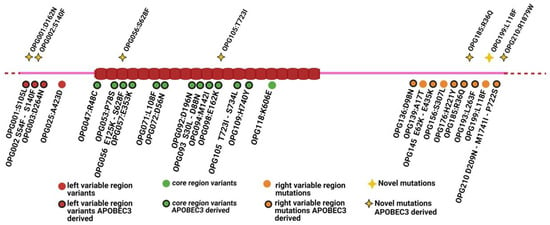
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}