
Dinaciclib, a Bimodal Agent Effective against Endometrial Cancer

 ,
,  ,
,  and
and 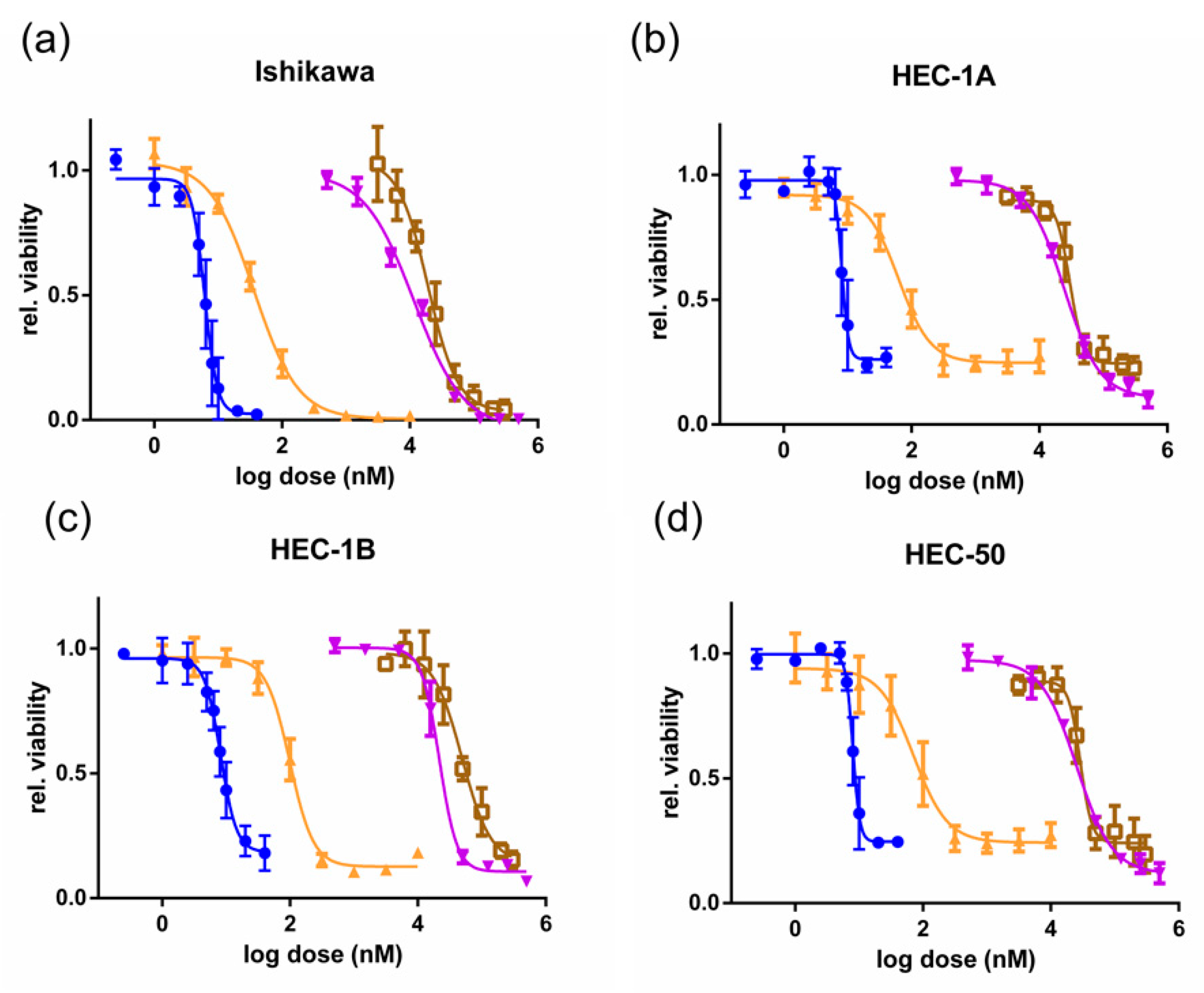{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Isolation and Expansion of Patient Tumor-Derived Cells
2.3. Cell Viability
2.4. Trypan Blue Live/Dead Cell Assay
2.5. RealTime-Glo Annexin V Apoptosis and Necrosis Assay
2.6. Cell Cycle Analysis
2.7. Immunoblotting
2.8. qPCR
2.9. Statistical Analysis
3. Results
3.1. CDKis Inhibit Growth in EC Cell Lines and Primary Cells
3.2. Dinaciclib Is Cytotoxic to Ishikawa Cells and Blocks the Proliferation of HEC-1A Cells
3.3. Dinaciclib Induces Cell Cycle Arrest
3.4. Dinaciclib Inhibits Phosphorylation of Pol II CTD at Ser2 and Reduces Expression of Anti-Apoptotic Genes
3.5. Dinaciclib Sensitizes EC Cell Lines to Cisplatin Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2015, 6736, 1–15. [Google Scholar] [CrossRef]
- Shen, F.; Gao, Y.; Ding, J.; Chen, Q. Is the positivity of estrogen receptor or progesterone receptor different between type 1 and type 2 endometrial cancer? Oncotarget 2017, 8, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Lax, S.F. Pathology of endometrial carcinoma. Adv. Exp. Med. Biol. 2017, 943, 75–96. [Google Scholar] [CrossRef]
- Burke, W.M.; Orr, J.; Leitao, M.; Salom, E.; Gehrig, P.; Olawaiye, A.B.; Brewer, M.; Boruta, D.; Villella, J.; Herzog, T.; et al. Endometrial cancer: A review and current management strategies: Part I. Gynecol. Oncol. 2014, 134, 385–392. [Google Scholar] [CrossRef]
- Amant, F.; Mirza, M.R.; Koskas, M.; Creutzberg, C.L. Cancer of the corpus uteri. Int. J. Gynecol. Obstet. 2018, 143, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passarello, K.; Kurian, S.; Villanueva, V. Endometrial Cancer: An Overview of Pathophysiology, Management, and Care. Semin. Oncol. Nurs. 2019, 35, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Dong, P.; Ihira, K.; Kudo, M.; Watari, H. Molecular-targeted therapies and precision medicine for endometrial cancer. Jpn. J. Clin. Oncol. 2019, 49, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.-M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Bonelli, M.; La Monica, S.; Fumarola, C.; Alfieri, R. Multiple effects of CDK4/6 inhibition in cancer: From cell cycle arrest to immunomodulation. Biochem. Pharmacol. 2019, 170. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Poornima, P.; Milani, M.; Gowda, K.; Amin, S.; Wang, H.G.; Cohen, G.M. Maritoclax and dinaciclib inhibit MCL-1 activity and induce apoptosis in both a MCL-1-dependent and -independent manner. Oncotarget 2015, 6, 12668–12681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009, 113, 4637–4645. [Google Scholar] [CrossRef] [Green Version]
- Shirsath, N.P.; Manohar, S.M.; Joshi, K.S. P276-00, a cyclin-dependent kinase inhibitor, modulates cell cycle and induces apoptosis in both in vitro and in vivo mantle cell lymphoma cell lines. Mol. Cancer 2012, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Marra, A.; Curigliano, G. Are all cyclin-dependent kinases 4/6 inhibitors created equal? NPJ Breast Cancer 2019, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Richards, D.A.; Edenfield, W.J.; Warner, S.L.; Mouritsen, L.; Bishop, R.; Anthony, S.P.; Bearss, D.; Vogelzang, N.J.; Whatcott, C. A phase I, first-in-human, open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study of oral TP-1287 administered daily to patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3611. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Do, K.T.; Tolaney, S.M.; Hilton, J.F.; Cleary, J.M.; Wolanski, A.; Beardslee, B.; Hassinger, F.; Bhushan, K.; Cai, D.; et al. Abstract CT047: Phase 1 dose-escalation study of the CDK inhibitor dinaciclib in combination with the PARP inhibitor veliparib in patients with advanced solid tumors. In Proceedings of the Clinical Trials, Washington, DC, USA, 1–5 April 2017; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2017; Volume 77, p. CT047. [Google Scholar]
- Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of cyclin-dependent kinase inhibitors in endometrial cancer. Int. J. Mol. Sci. 2019, 20, 2353. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, J.; Kamata, Y.; Seo, N.; Okayasu, I.; Kuramoto, H. Stimulatory effect of estrogen on the growth of endometrial cancer cells is regulated by cell-cycle regulators. J. Steroid Biochem. Mol. Biol. 2007, 107, 163–171. [Google Scholar] [CrossRef]
- Che, Q.; Xiao, X.; Xu, J.; Liu, M.; Lu, Y.; Liu, S.; Dong, X. 17β-estradiol promotes endometrial cancer proliferation and invasion through IL-6 pathway. Endocr. Connect. 2019, 8, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Rižner, T.L. Estrogen biosynthesis, phase I and phase II metabolism, and action in endometrial cancer. Mol. Cell. Endocrinol. 2013, 381, 124–139. [Google Scholar] [CrossRef]
- Markowska, A.; Pawałowska, M.; Lubin, J.; Markowska, J. Signalling pathways in endometrial cancer. Wspolczesna Onkol. 2014, 18, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Mu, K.; Wang, Y.; Zhou, Z.; Zhang, J.; Sheng, Y.; Zhang, T. CyclinD1, a prominent prognostic marker for endometrial diseases. Diagn. Pathol. 2013, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, A.; Sebo, T.J.; Cliby, W.A.; Keeney, G.L.; Riehle, D.L.; Lesnick, T.G.; Podratz, K.C. Role of bcl-2 in endometrioid corpus cancer: An experimental study. Anticancer Res. 2006, 26, 823–827. [Google Scholar] [PubMed]
- Witek, Ł.; Janikowski, T.; Bodzek, P.; Olejek, A.; Mazurek, U. Expression of tumor suppressor genes related to the cell cycle in endometrial cancer patients. Adv. Med. Sci. 2016, 61, 317–324. [Google Scholar] [CrossRef]
- Bell, D.W.; Ellenson, L.H. Molecular Genetics of Endometrial Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 339–367. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, H.H.; Pharma, A.; Gmbh, D.; Biotechnology, C.; Box, P.O. Mechanisms of Action of Flavopiridol. Crit. Rev. Oncol. Hematol. 2001, 38, 139–170. [Google Scholar] [CrossRef]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2354. [Google Scholar] [CrossRef] [Green Version]
- Margarit, L.; Taylor, A.; Roberts, M.H.; Hopkins, L.; Davies, C.; Brenton, A.G.; Conlan, R.S.; Bunkheila, A.; Joels, L.; White, J.O.; et al. MUC1 as a discriminator between endometrium from fertile and infertile patients with PCOS and endometriosis. J. Clin. Endocrinol. Metab. 2010, 95, 5320–5329. [Google Scholar] [CrossRef] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.V.; Chambers, S.H.; Smith, P.J. A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 1987, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, X.; Liu, C.; Ren, M.; Gao, S.; Zhao, G.; Zhang, T.; Yang, Q. Validation of reference genes for the normalization of RT-qPCR expression studies in human tongue carcinoma cell lines and tissue. Oncol. Lett. 2017, 13, 3951–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.K.; Huang, S.L.; Chang, T.C.; Chao, C.C.K. Sorafenib induces endometrial carcinoma apoptosis by inhibiting Elk-1-dependent Mcl-1 transcription and inducing Akt/GSK3β-dependent protein degradation. J. Cell. Biochem. 2013, 114, 1819–1831. [Google Scholar] [CrossRef]
- Eimani, B.G.; Sanati, M.H.; Houshmand, M.; Ataei, M.; Akbarian, F.; Shakhssalim, N. Expression and prognostic significance of Bcl-2 and Bax in the progression and clinical outcome of transitional bladder cell carcinoma. Cell J. 2014, 15, 356–363. [Google Scholar]
- Chen, X.Q.; Yang, S.; Kang, M.Q.; Li, Z.Y.; Lu, H.S.; Lin, T.Y. Survivin expression in human lung cancer and the influence of its downregulation on the biological behavior of human lung cancer cells. Exp. Ther. Med. 2012, 3, 1010–1014. [Google Scholar] [CrossRef]
- Rouette, A.; Parent, S.; Girouard, J.; Leblanc, V.; Asselin, E. Cisplatin increases B-cell-lymphoma-2 expression via activation of protein kinase C and Akt2 in endometrial cancer cells. Int. J. Cancer 2012, 130, 1755–1767. [Google Scholar] [CrossRef]
- Albert, A.; Lavoie, S.; Vincent, M. A hyperphosphorylated form of RNA polymerase II is the major interphase antigen of the phosphoprotein antibody MPM-2 and interacts with the peptidyl-prolyl isomerase Pin1. J. Cell Sci. 1999, 112, 2493–2500. [Google Scholar]
- Booher, R.N.; Hatch, H.; Dolinski, B.M.; Nguyen, T.; Harmonay, L.; Al-Assaad, A.-S.; Ayers, M.; Nebozhyn, M.; Loboda, A.; Hirsch, H.A.; et al. MCL1 and BCL-xL Levels in Solid Tumors Are Predictive of Dinaciclib-Induced Apoptosis. PLoS ONE 2014, 9, e108371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, G.P.; Hogg, S.J.; Kats, L.M.; Vidacs, E.; Baker, A.J.; Gilan, O.; Lefebure, M.; Martin, B.P.; Dawson, M.A.; Johnstone, R.W.; et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2015, 29, 1437–1441. [Google Scholar] [CrossRef]
- Chen, X.-X.; Xie, F.-F.; Zhu, X.-J.; Lin, F.; Pan, S.-S.; Gong, L.-H.; Qiu, J.-G.; Zhang, W.-J.; Jiang, Q.-W.; Mei, X.-L.; et al. Cyclin-dependent kinase inhibitor dinaciclib potently synergizes with cisplatin in preclinical models of ovarian cancer. Oncotarget 2015, 6, 14926–14939. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, L.; Zhang, J.; Cai, W.; Zhao, S.; Liu, S. Accumulated cytotoxicity of CDK inhibitor dinaciclib with first-line chemotherapy drugs in salivary adenoid cystic carcinoma cells. Odontology 2020, 108, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Y.; Zhang, C.; Chu, J.; Wu, Y.; Li, Y.; Liu, J.; Li, Q.; Li, S.; Shi, Q.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Flynn, J.; Jones, J.; Johnson, A.J.; Andritsos, L.; Maddocks, K.; Jaglowski, S.; Hessler, J.; Grever, M.R.; Im, E.; Zhou, H.; et al. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 2015, 29, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, Z.; Pang, J.C.; Yu, Y.; Bieerkehazhi, S.; Lu, J.; Hu, T.; Zhao, Y.; Xu, X.; Zhang, H.; et al. Multiple CDK inhibitor dinaciclib suppresses neuroblastoma growth via inhibiting CDK2 and CDK9 activity. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, C.; Wang, X.; Becker, D. Expression analysis and molecular targeting of cyclin-dependent kinases in advanced melanoma: Functional analysis and molecular targeting of cyclin-dependent kinase family members in advanced melanoma. Cell Cycle 2011, 10, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, S.; Khera, N.; Guo, Z.; Hoog, J.; Li, S.; Ma, C.X. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget 2016, 7, 56864–56875. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.F.; Der Lin, J.; Hsueh, C.; Chou, T.C.; Wong, R.J. A cyclin-dependent kinase inhibitor, dinaciclib in preclinical treatment models of thyroid cancer. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Fischer, P.M. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef] [PubMed]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Hu, L.; Wu, Z.; Chen, Z.; Liu, S.; Xu, X.; Qian, A. CDK12: A Potent Target and Biomarker for Human Cancer Therapy. Cells 2020, 9, 1483. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howard, D.; James, D.; Murphy, K.; Garcia-Parra, J.; Pan-Castillo, B.; Rex, S.; Moul, A.; Jones, E.; Bilbao-Asensio, M.; Michue-Seijas, S.; et al. Dinaciclib, a Bimodal Agent Effective against Endometrial Cancer. Cancers 2021, 13, 1135. https://doi.org/10.3390/cancers13051135
Howard D, James D, Murphy K, Garcia-Parra J, Pan-Castillo B, Rex S, Moul A, Jones E, Bilbao-Asensio M, Michue-Seijas S, et al. Dinaciclib, a Bimodal Agent Effective against Endometrial Cancer. Cancers. 2021; 13(5):1135. https://doi.org/10.3390/cancers13051135
Chicago/Turabian StyleHoward, David, David James, Kate Murphy, Jezabel Garcia-Parra, Belen Pan-Castillo, Stuart Rex, Annemarie Moul, Eilir Jones, Marc Bilbao-Asensio, Saul Michue-Seijas, and et al. 2021. "Dinaciclib, a Bimodal Agent Effective against Endometrial Cancer" Cancers 13, no. 5: 1135. https://doi.org/10.3390/cancers13051135