
Disruption of Metapopulation Structure Reduces Tasmanian Devil Facial Tumour Disease Spread at the Expense of Abundance and Genetic Diversity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
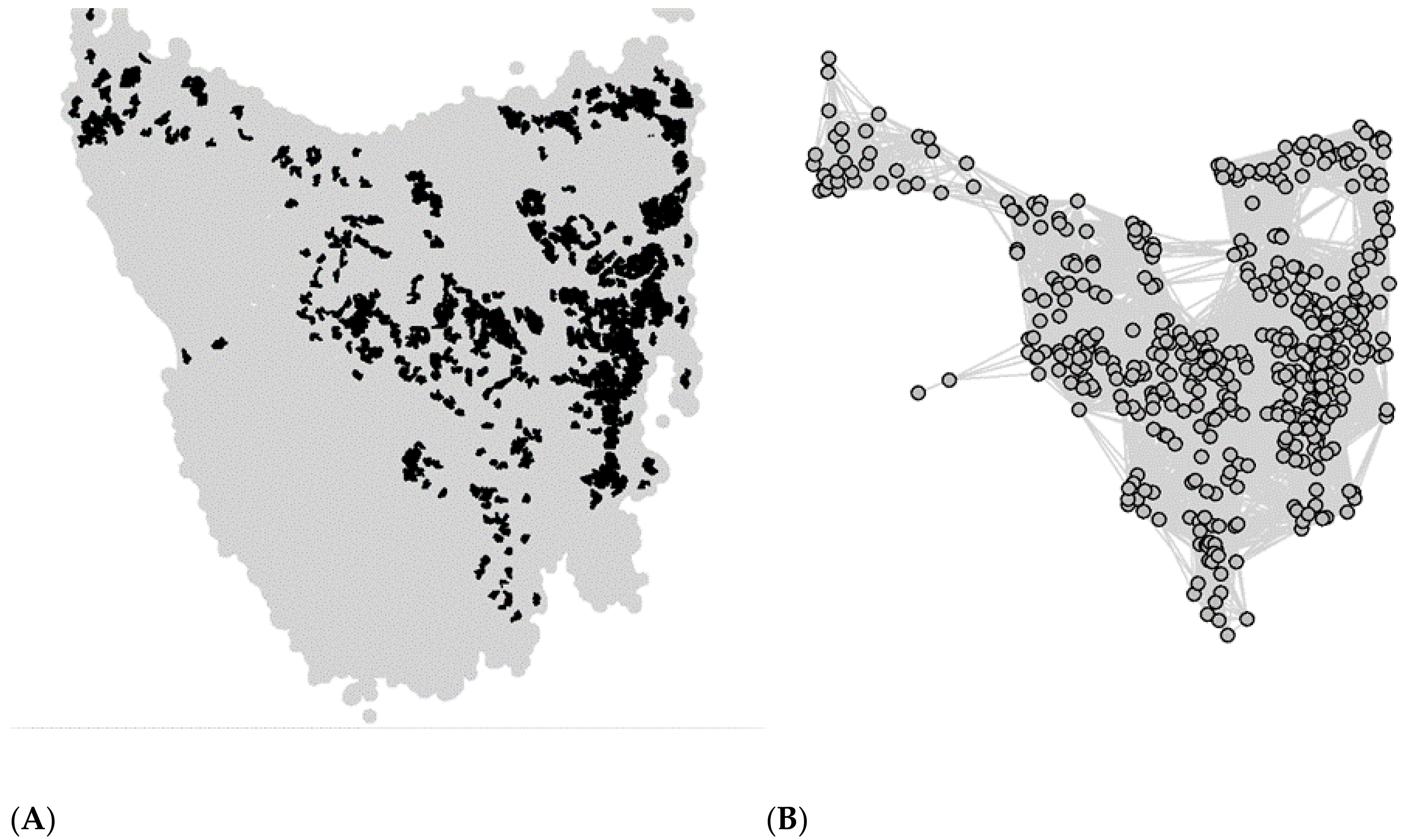
2.1. Metapopulation Structure
2.2. Individual-Based Model
2.3. Tracing the Mixing of Individuals from Different Populations
2.4. Model Simulations
2.5. Metapopulation Analysis
- (1)
- Total metapopulation size: quantified as the median, across the last 520 weeks (ten years) of the simulation, of the sum of the sizes (i.e., number of individuals) of all local populations at each time step.
- (2)
- Proportion of local populations where at least one case of DFTD was recorded during the entire simulated time period.
2.6. Pattern Matching of Disease Spread to Empirical Data
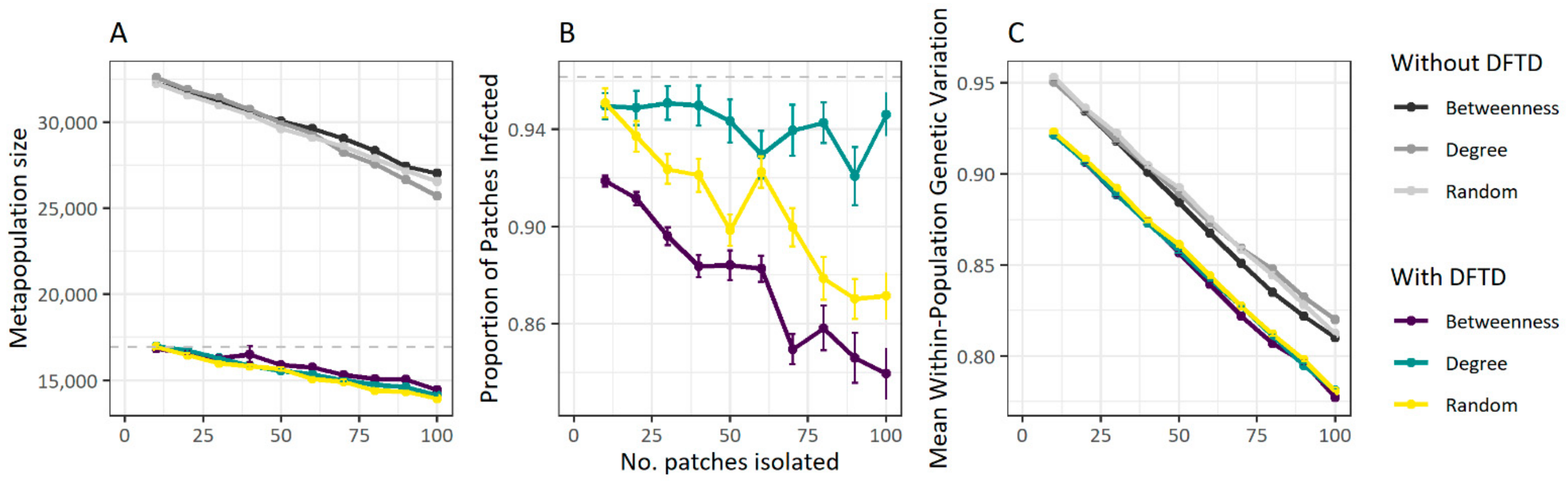
2.7. Metapopulation Fragmentation Experiments
- (1)
- Random: Populations to be isolated were randomly chosen.
- (2)
- Degree: Isolated populations were chosen in order according to their degree (i.e., the number of connections of the local population), from the most connected to the least connected population.
- (3)
- Betweenness: Populations to isolate were selected in order according to their betweenness centrality, a node-level network measure that quantifies the extent to which the shortest paths connecting any two nodes (populations) in the network (the metapopulation) comprise the focal node. Local populations were thus ranked from most to least central according to their betweenness centrality, and those with the highest centrality scores were removed first. Betweenness centrality of a local population v was calculated thus [48]
3. Results
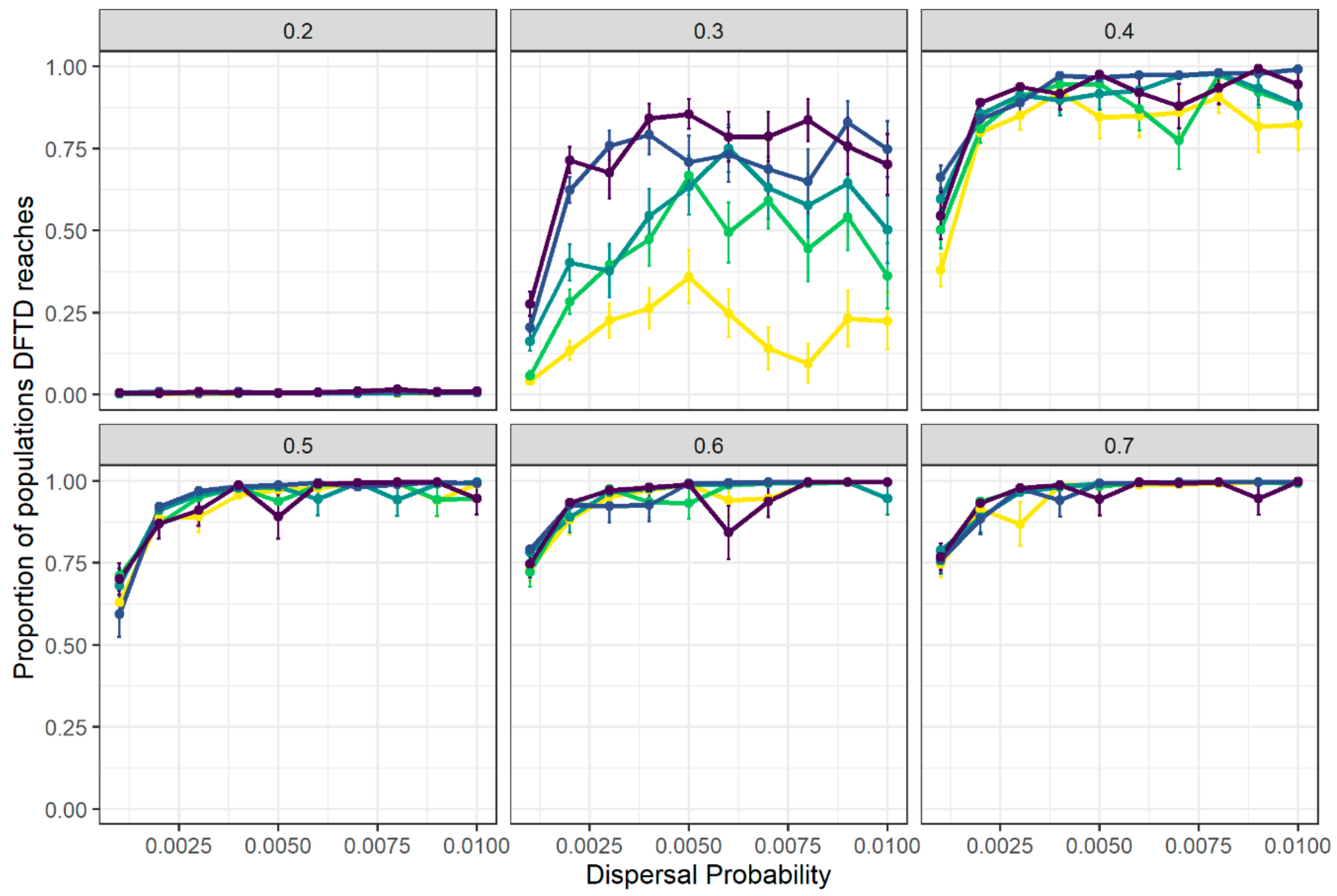
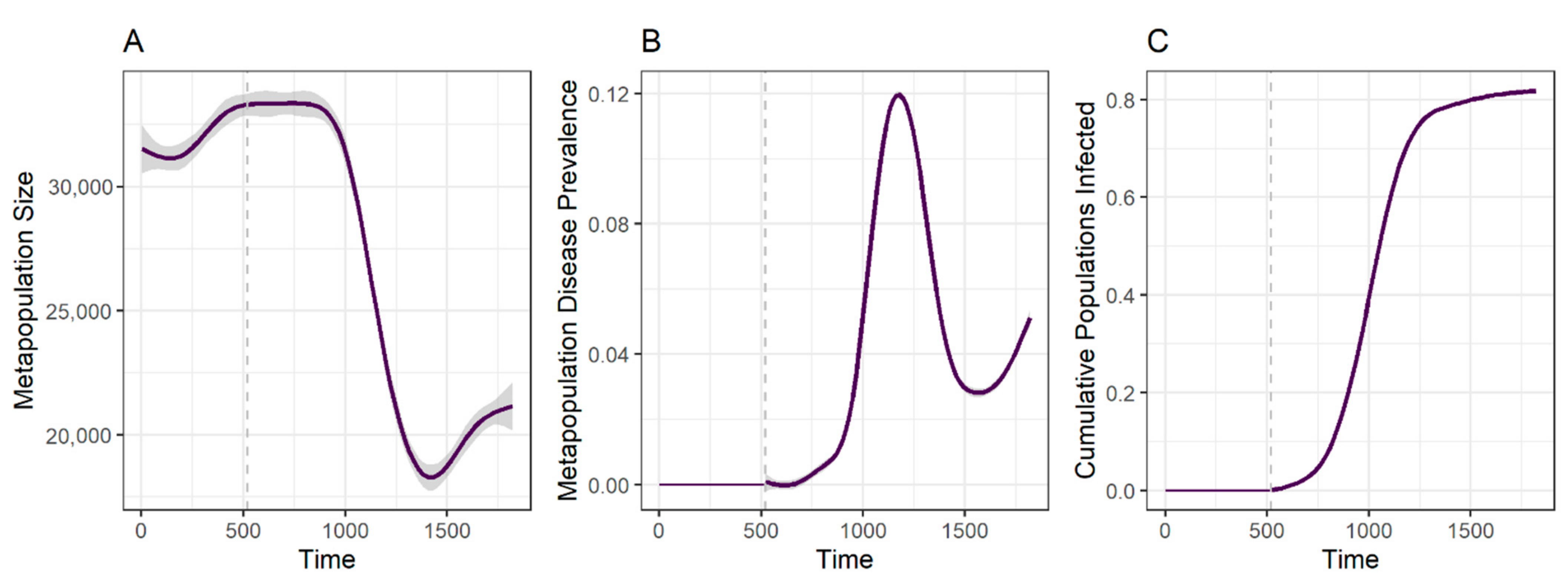
3.1. Disease Spread Is Driven by the Interplay between Local Transmission and Regional Movement
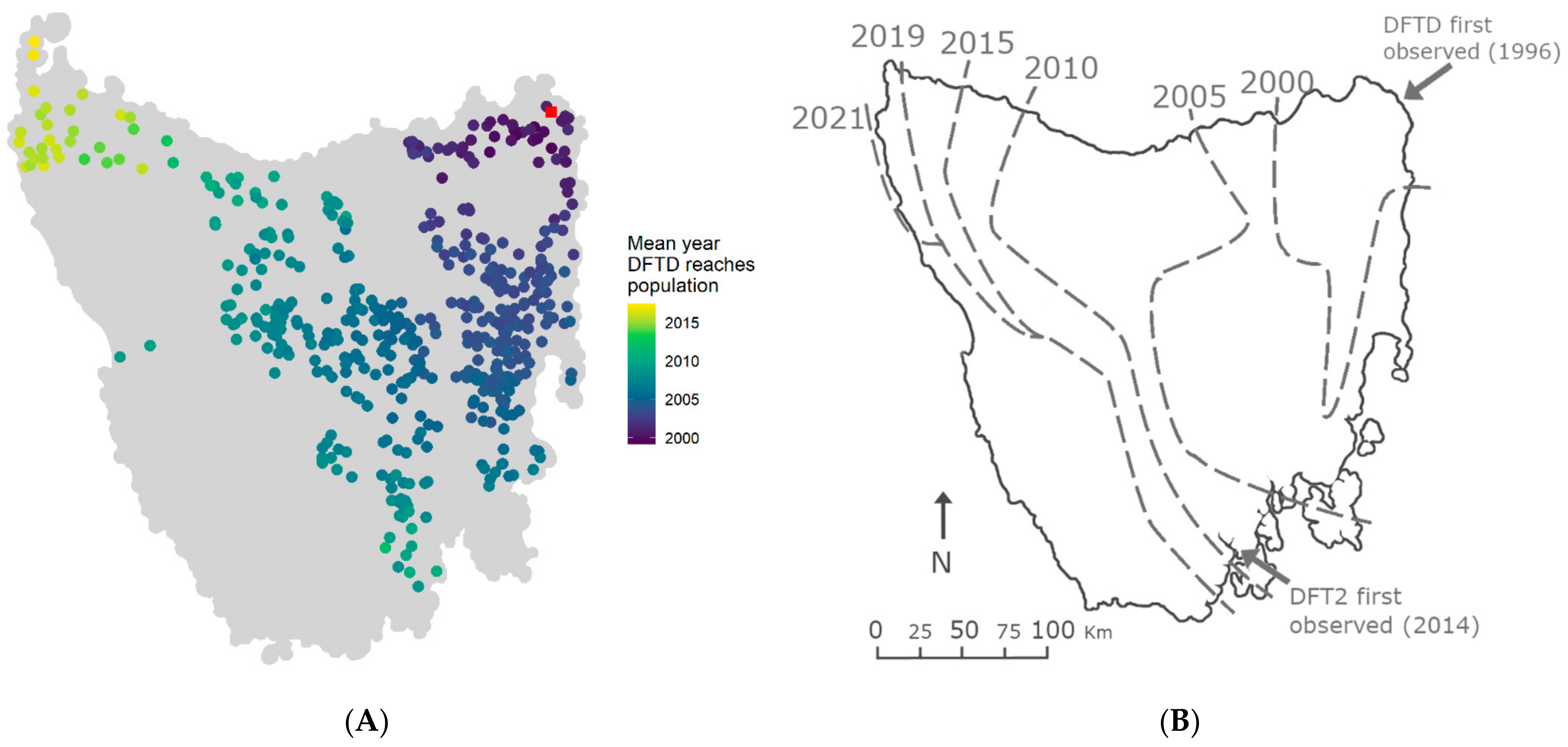
3.2. Spatial Spread of DFTD across Tasmania Is Explained by Metapopulation Dynamics
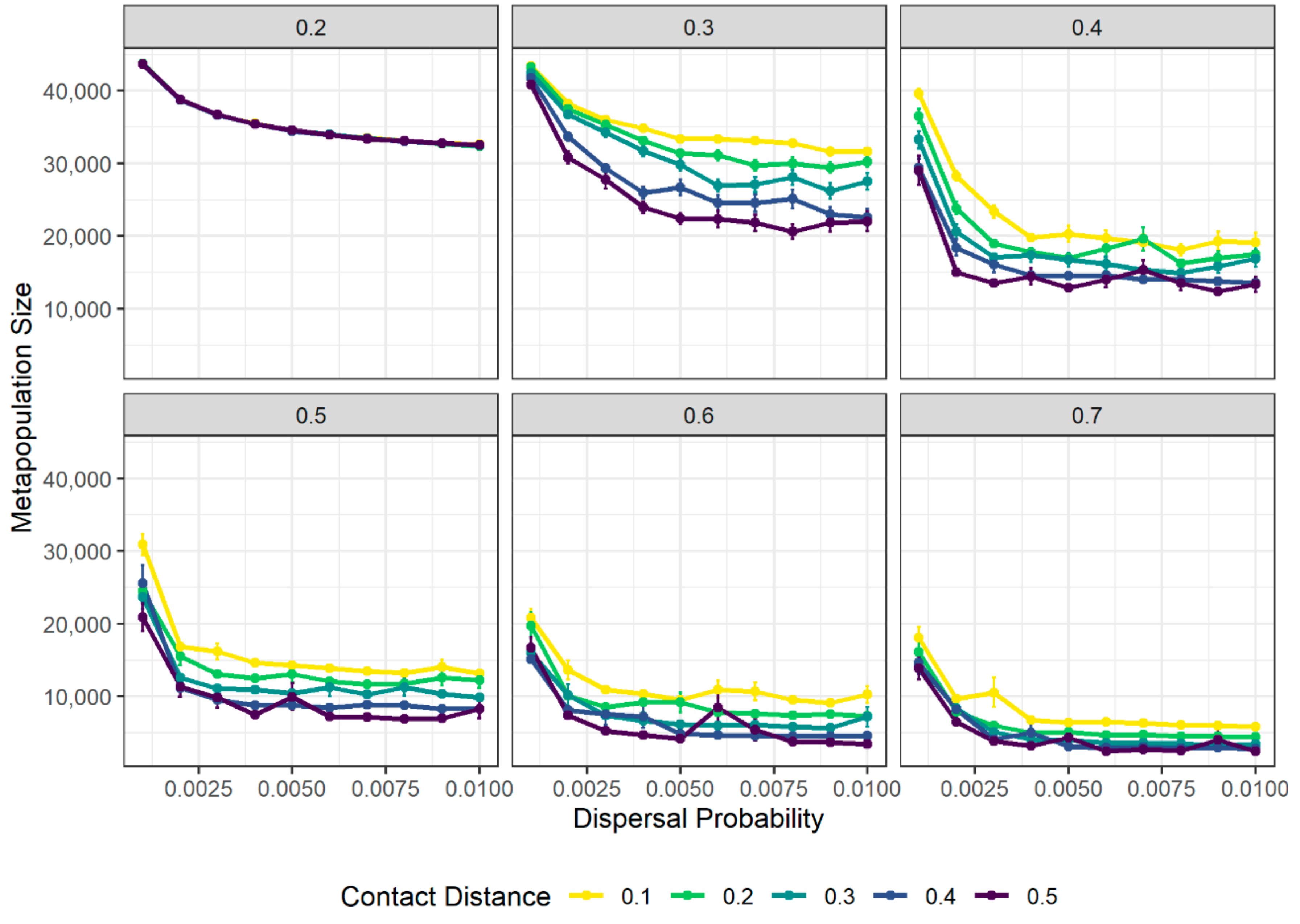
3.3. Fragmentation Reduces Disease Spread at the Cost of Population Size
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Castro, F.; Bolker, B. Mechanisms of disease-induced extinction. Ecol. Lett. 2005, 8, 117–126. [Google Scholar] [CrossRef]
- McAloose, D.; Newton, A.L. Wildlife cancer: A conservation perspective. Nat. Rev. Cancer 2009, 9, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Hamede, R.; Owen, R.; Siddle, H.; Peck, S.; Jones, M.; Dujon, A.; Giraudeau, M.; Roche, B.; Ujvari, B.; Thomas, F. The ecology and evolution of wildlife cancers: Applications for management and conservation. Evol. Appl. 2020, 13, 1719–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraudeau, M.; Sepp, T.; Ujvari, B.; Ewald, P.W.; Thomas, F. Human activities might influence oncogenic processes in wild animal populations. Nat. Ecol. Evol. 2018, 2, 1065–1070. [Google Scholar] [CrossRef]
- Pesavento, P.A.; Agnew, D.; Keel, M.K.; Woolard, K.D. Cancer in wildlife: Patterns of emergence. Nat. Rev. Cancer 2018, 18, 646–661. [Google Scholar] [CrossRef]
- Ujvari, B.; Klaassen, M.; Raven, N.; Russell, T.; Vittecoq, M.; Hamede, R.; Thomas, F.; Madsen, T. Genetic diversity, inbreeding and cancer. Proc. R Soc. B Biol. Sci. 2018, 285, 20172589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meslin, F.X. Global aspects of emerging and potential zoonoses: A WHO perspective. Emerg Infect. Dis 1997, 3, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Morales, A.J.; Bonilla-Aldana, D.K.; Balbin-Ramon, G.J.; Rabaan, A.A.; Sah, R.; Paniz-Mondolfi, A.; Pagliano, P.; Esposito, S. History is repeating itself: Probable zoonotic spillover as the cause of the 2019 novel Coronavirus Epidemic. Infez. Med. 2020, 28, 3–5. [Google Scholar] [PubMed]
- Grenfell, B.T.; Gulland, F.M.D. Introduction: Ecological impact of parasitism on wildlife host populations. Parasitology 1995, 111, S3–S14. [Google Scholar] [CrossRef]
- Daszak, P.; Cunningham, A.A.; Hyatt, A.D. Emerging Infectious Diseases of Wildlife-- Threats to Biodiversity and Human Health. Science 2000, 287, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Wobeser, G. Disease management strategies for wildlife. Rev. Sci. Tech. OIE 2002, 21, 159–178. [Google Scholar] [CrossRef] [PubMed]
- May, R.M.; Anderson, R.M. Population biology of infectious diseases: Part II. Nature 1979, 280, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Colizza, V.; Vespignani, A. Epidemic modeling in metapopulation systems with heterogeneous coupling pattern: Theory and simulations. J. Theor. Biol. 2008, 251, 450–467. [Google Scholar] [CrossRef] [Green Version]
- North, A.R.; Godfray, H.C.J. The dynamics of disease in a metapopulation: The role of dispersal range. J. Theor. Biol. 2017, 418, 57–65. [Google Scholar] [CrossRef]
- Wilson, M.E. Travel and the emergence of infectious diseases. Emerg. Infect. Dis 1995, 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Fèvre, E.M.; de Bronsvoort, B.M.C.; Hamilton, K.A.; Cleaveland, S. Animal movements and the spread of infectious diseases. Trends Microbiol. 2006, 14, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Dorling, D.; Cao, G.; Liu, T. Changes in population movement make COVID-19 spread differently from SARS. Soc. Sci. Med. 2020, 255, 113036. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.; Lurgi, M.; Collins, B.; Lucini, B.; Kao, R.R.; Lloyd, A.L.; Frost, S.D.W.; Gravenor, M.B. Disease control across urban–rural gradients. J. R. Soc. Interface 2020, 17, 20200775. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, C.; Shen, W.; Tang, F.; Lei, H.; Xie, Y.; Cao, Z.; Tang, K.; Bai, J.; Xiao, L.; et al. Impact of population movement on the spread of 2019-nCoV in China. Emerg. Microbes Infect. 2020, 9, 988–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viboud, C.; Bjørnstad, O.N.; Smith, D.L.; Simonsen, L.; Miller, M.A.; Grenfell, B.T. Synchrony, Waves, and Spatial Hierarchies in the Spread of Influenza. Science 2006, 312, 447–451. [Google Scholar] [CrossRef] [Green Version]
- Hess, G. Disease in Metapopulation Models: Implications for Conservation. Ecology 1996, 77, 1617–1632. [Google Scholar] [CrossRef]
- Okano, J.T.; Sharp, K.; Valdano, E.; Palk, L.; Blower, S. HIV transmission and source–sink dynamics in sub-Saharan Africa. Lancet HIV 2020, 7, e209–e214. [Google Scholar] [CrossRef]
- Watkinson, A.R.; Sutherland, W.J. Sources, Sinks and Pseudo-Sinks. J. Anim. Ecol. 1995, 64, 126–130. [Google Scholar] [CrossRef]
- Robinson, S.J.; Samuel, M.D.; Rolley, R.E.; Shelton, P. Using landscape epidemiological models to understand the distribution of chronic wasting disease in the Midwestern USA. Landsc. Ecol. 2013, 28, 1923–1935. [Google Scholar] [CrossRef]
- Fulford, G.R.; Roberts, M.G.; Heesterbeek, J.A.P. The Metapopulation Dynamics of an Infectious Disease: Tuberculosis in Possums. Theor. Popul. Biol. 2002, 61, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, C.; Baars, C.; Hesterman, H.; Hocking, G.; Jones, M.; Lazenby, B.; Mann, D.; Mooney, N.; Pemberton, D.; Pyecroft, S.; et al. Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol. Conserv. 2006, 131, 307–324. [Google Scholar] [CrossRef]
- Cunningham, C.X.; Comte, S.; McCallum, H.; Hamilton, D.G.; Hamede, R.; Storfer, A.; Hollings, T.; Ruiz-Aravena, M.; Kerlin, D.H.; Brook, B.W.; et al. Quantifying 25 years of disease-caused declines in Tasmanian devil populations: Host density drives spatial pathogen spread. Ecol. Lett. 2021, 24, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Hamede, R.K.; McCallum, H.; Jones, M. Biting injuries and transmission of Tasmanian devil facial tumour disease. J. Anim. Ecol. 2013, 82, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Siddle, H.V.; Kreiss, A.; Tovar, C.; Yuen, C.K.; Cheng, Y.; Belov, K.; Swift, K.; Pearse, A.-M.; Hamede, R.; Jones, M.; et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl. Acad. Sci. 2013, 110, 5103–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallum, H.; Jones, M.; Hawkins, C.; Hamede, R.; Lachish, S.; Sinn, D.L.; Beeton, N.; Lazenby, B. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology 2009, 90, 3379–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamede, R.K.; Bashford, J.; McCallum, H.; Jones, M. Contact networks in a wild Tasmanian devil (Sarcophilus harrisii) population: Using social network analysis to reveal seasonal variability in social behaviour and its implications for transmission of devil facial tumour disease. Ecol. Lett. 2009, 12, 1147–1157. [Google Scholar] [CrossRef]
- Hamilton, D.G.; Jones, E.M.; Cameron, E.Z.; McCallum, H.; Storfer, A.; Hohenlohe, A.P.; Hamede, R. Rate of intersexual interactions affects injury likelihood in Tasmanian devil contact networks. Behav. Ecol. 2019, 30, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Lazenby, B.T.; Tobler, M.W.; Brown, W.E.; Hawkins, C.E.; Hocking, G.J.; Hume, F.; Huxtable, S.; Iles, P.; Jones, M.; Lawrence, C.; et al. Density trends and demographic signals uncover the long-term impact of transmissible cancer in Tasmanian devils. J. Appl. Ecol. 2018, 55, 1368–1379. [Google Scholar] [CrossRef]
- McCallum, H.; Tompkins, D.M.; Jones, M.; Lachish, S.; Marvanek, S.; Lazenby, B.; Hocking, G.; Wiersma, J.; Hawkins, C.E. Distribution and Impacts of Tasmanian Devil Facial Tumor Disease. EcoHealth 2007, 4, 318. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.E.; Cockburn, A.; Hamede, R.; Hawkins, C.; Hesterman, H.; Lachish, S.; Mann, D.; McCallum, H.; Pemberton, D. Life-history change in disease-ravaged Tasmanian devil populations. Proc. Natl. Acad. Sci. USA 2008, 105, 10023–10027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeton, N.; McCallum, H. Models predict that culling is not a feasible strategy to prevent extinction of Tasmanian devils from facial tumour disease. J. Anim. Ecol. 2012, 48, 1315–1323. [Google Scholar] [CrossRef]
- Bruno, C.D.; Comar, T.; Powell, M.O.; Tameklo, A. Age-Structured and Vaccination Models of Devil Facial Tumor Disease. Spora J. Biomath 2017, 3. [Google Scholar] [CrossRef]
- Wells, K.; Hamede, R.K.; Jones, M.E.; Hohenlohe, P.A.; Storfer, A.; McCallum, H.I. Individual and temporal variation in pathogen load predicts long-term impacts of an emerging infectious disease. Ecology 2019, 100, e02613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, V.; Eriksson, A.; Mehlig, B.; Manica, A. A metapopulation model of the spread of the Devil Facial Tumour Disease predicts the long term collapse of its host but not its extinction. arXiv 2018, arXiv:1806.05449. [Google Scholar] [CrossRef]
- Hamede, R.K.; Pearse, A.-M.; Swift, K.; Barmuta, L.A.; Murchison, E.P.; Jones, M.E. Transmissible cancer in Tasmanian devils: Localized lineage replacement and host population response. Proc. R Soc. B Biol. Sci. 2015, 282, 20151468. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.; Hamede, R.K.; Kerlin, D.H.; Storfer, A.; Hohenlohe, P.A.; Jones, M.E.; McCallum, H.I. Infection of the fittest: Devil facial tumour disease has greatest effect on individuals with highest reproductive output. Ecol. Lett. 2017, 20, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Department of the Environment. Sarcophilus harrisii in Species Profile and Threats Database, Department of the Environment, Canberra 2020. Available online: http://www.environment.gov.au/sprat (accessed on 19 June 2020).
- Guiler, E.R. Obsevations on the Tasmanian Devil, Sarcophilus harrisii (Marsupialia: Dasyuridae) I. Numbers, home, range, movements and food in two populations. Aust. J. Zool. 1970, 18, 49–62. [Google Scholar] [CrossRef]
- Lachish, S.; Miller, K.J.; Storfer, A.; Goldizen, A.W.; Jones, M.E. Evidence that disease-induced population decline changes genetic structure and alters dispersal patterns in the Tasmanian devil. Heredity 2011, 106, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Department of Primary Industries, Parks, Water and Environment (Tasmania). TASVEG 2.0. 2009. Available online: https://www.thelist.tas.gov.au/app/content/data/geo-meta-data-record?detailRecordUID=99de4ddb-d9c3-430b-ad29-055b80cc46ec (accessed on 17 June 2020).
- Nei, M. Analysis of Gene Diversity in Subdivided Populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.X.; Johnson, C.N.; Hollings, T.; Kreger, K.; Jones, M.E. Trophic rewilding establishes a landscape of fear: Tasmanian devil introduction increases risk-sensitive foraging in a key prey species. Ecography 2019, 42, 2053–2059. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1979, 1, 215–239. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R. A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Yang, H.-X.; Wang, B.-H. Immunization of traffic-driven epidemic spreading. Phys. Stat. Mech. Appl. 2016, 443, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Satorras, R.; Vespignani, A. Immunization of complex networks. Phys. Rev. E 2002, 65, 036104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.H.; Kodric-Brown, A. Turnover Rates in Insular Biogeography: Effect of Immigration on Extinction. Ecology 1977, 58, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Gotelli, N.J. Metapopulation Models: The Rescue Effect, the Propagule Rain, and the Core-Satellite Hypothesis. Am. Nat. 1991, 138, 768–776. [Google Scholar] [CrossRef]
- Hess, G.R. Conservation Corridors and Contagious Disease: A Cautionary Note. Conserv. Biol. 1994, 8, 256–262. [Google Scholar] [CrossRef]
- JJousimo, J.; Tack, A.J.M.; Ovaskainen, O.; Mononen, T.; Susi, H.; Tollenaere, C.; Laine, A.-L. Ecological and evolutionary effects of fragmentation on infectious disease dynamics. Science 2014, 344, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Levins, R. Some Demographic and Genetic Consequences of Environmental Heterogeneity for Biological Control. Bull. Entomol. Soc. Am. 1969, 15, 237–240. [Google Scholar] [CrossRef]
- Brochier, B.; Kieny, M.P.; Costy, F.; Coppens, P.; Bauduin, B.; Lecocq, J.P.; Languet, B.; Chappuis, G.; Desmettre, P.; Afiademanyo, K.; et al. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature 1991, 354, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Pye, R.; Kreiss, A.; Cheng, Y.; Brown, G.K.; Darby, J.; Malley, R.C.; Siddle, H.V.T.; Skjødt, K.; Kaufman, J.; et al. Regression of devil facial tumour disease following immunotherapy in immunised Tasmanian devils. Sci. Rep. 2017, 7, srep43827. [Google Scholar] [CrossRef]
- Pye, R.; Patchett, A.; McLennan, E.; Thomson, R.; Carver, S.; Fox, S.; Pemberton, D.; Kreiss, A.; Morelli, A.B.; Silva, A.; et al. Immunization Strategies Producing a Humoral IgG Immune Response against Devil Facial Tumor Disease in the Majority of Tasmanian Devils Destined for Wild Release. Front. Immunol. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.S.; Siddle, H.V. Devil Facial Tumours: Towards a Vaccine. Immunol. Invest. 2019, 48, 719–736. [Google Scholar] [CrossRef]
- Gandon, S.; Mackinnon, M.J.; Nee, S.; Read, A.F. Imperfect vaccines and the evolution of pathogen virulence. Nature 2001, 414, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.; Walkden-Brown, S.W.; Nair, V. Imperfect Vaccination Can Enhance the Transmission of Highly Virulent Pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; McCallum, H.I.; Jones, M.E.; Lawrance, M.F.; Hamede, R.K.; Storfer, A. Conserving adaptive potential: Lessons from Tasmanian devils and their transmissible cancer. Conserv Genet. 2019, 20, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Hamede, R.; Madsen, T.; McCallum, H.; Storfer, A.; Hohenlohe, P.A.; Siddle, H.; Kaufman, J.; Giraudeau, M.; Jones, M.; Thomas, F.; et al. Darwin, the devil, and the management of transmissible cancers. Conserv. Biol. 2020, 35, 748–751. [Google Scholar] [CrossRef]
- Pye, R.; Hamede, R.; Siddle, H.V.; Caldwell, A.; Knowles, G.W.; Swift, K.; Kreiss, A.; Jones, M.; Lyons, A.; Woods, G.M. Demonstration of immune responses against devil facial tumour disease in wild Tasmanian devils. Biol. Lett. 2016, 12, 20160553. [Google Scholar] [CrossRef] [Green Version]
- Margres, M.J.; Ruiz-Aravena, M.; Hamede, R.; Jones, E.M.; Lawrance, M.F.; Hendricks, A.S.; Patton, A.; Davis, B.W.; Ostrander, A.E.; McCallum, H.; et al. The Genomic Basis of Tumor Regression in Tasmanian Devils (Sarcophilus harrisii). Genome Biol. Evol. 2018, 10, 3012–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, A.H.; Lawrance, M.F.; Margres, M.J.; Kozakiewicz, C.P.; Hamede, R.; Ruiz-Aravena, M.; Hamilton, D.G.; Comte, S.; Ricci, L.E.; Taylor, R.L.; et al. A transmissible cancer shifts from emergence to endemism in Tasmanian devils. Science 2020, 370, eabb9772. [Google Scholar] [CrossRef]
- Pye, R.; Pemberton, D.; Tovar, C.; Tubio, J.; Dun, K.; Fox, S.; Darby, J.; Hayes, D.; Knowles, G.W.; Kreiss, A.; et al. A second transmissible cancer in Tasmanian devils. Proc. Natl. Acad. Sci. USA 2016, 113, 374–379. [Google Scholar] [CrossRef] [Green Version]
- James, S.; Jennings, G.; Kwon, Y.M.; Stammnitz, M.; Fraik, A.; Storfer, A.; Comte, S.; Pemberton, D.; Fox, S.; Brown, B.; et al. Tracing the rise of malignant cell lines: Distribution, epidemiology and evolutionary interactions of two transmissible cancers in Tasmanian devils. Evol. Appl. 2019, 12, 1772–1780. [Google Scholar] [CrossRef] [PubMed]
- Flies, A.S.; Flies, E.J.; Fox, S.; Gilbert, A.; Johnson, S.R.; Liu, G.-S.; Lyons, A.B.; Patchett, A.L.; Pemberton, D.; Pye, R.J. An oral bait vaccination approach for the Tasmanian devil facial tumor diseases. Expert. Rev. Vaccines 2020, 19, 1–10. [Google Scholar] [CrossRef]
- Zamberletti, P.; Zaffaroni, M.; Accatino, F.; Creed, I.F.; De Michele, C. Connectivity among wetlands matters for vulnerable amphibian populations in wetlandscapes. Ecol. Model. 2018, 384, 119–127. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durrant, R.; Hamede, R.; Wells, K.; Lurgi, M. Disruption of Metapopulation Structure Reduces Tasmanian Devil Facial Tumour Disease Spread at the Expense of Abundance and Genetic Diversity. Pathogens 2021, 10, 1592. https://doi.org/10.3390/pathogens10121592
Durrant R, Hamede R, Wells K, Lurgi M. Disruption of Metapopulation Structure Reduces Tasmanian Devil Facial Tumour Disease Spread at the Expense of Abundance and Genetic Diversity. Pathogens. 2021; 10(12):1592. https://doi.org/10.3390/pathogens10121592
Chicago/Turabian StyleDurrant, Rowan, Rodrigo Hamede, Konstans Wells, and Miguel Lurgi. 2021. "Disruption of Metapopulation Structure Reduces Tasmanian Devil Facial Tumour Disease Spread at the Expense of Abundance and Genetic Diversity" Pathogens 10, no. 12: 1592. https://doi.org/10.3390/pathogens10121592