


Viruses 2024, 16(5), 732; https://doi.org/10.3390/v16050732 (registering DOI) - 05 May 2024
Abstract
The L 1 region of bovine adenovirus (BAdV)-3 encodes a multifunctional protein named protein VII. Anti-protein VII sera detected a protein of 26 kDa in transfected or BAdV-3-infected cells, which localizes to nucleus and nucleolus of infected/transfected cells. Analysis of mutant protein VII
[...] Read more.
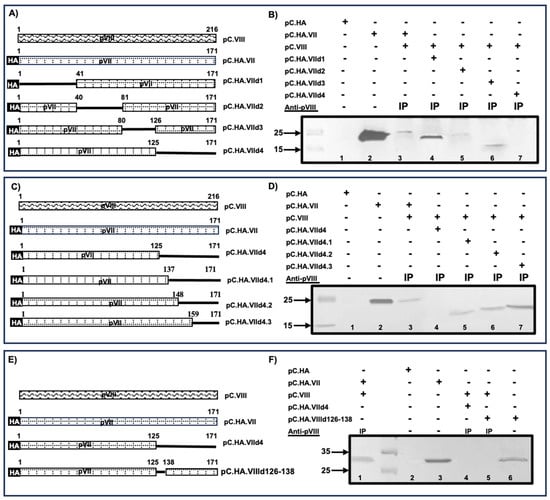
The L 1 region of bovine adenovirus (BAdV)-3 encodes a multifunctional protein named protein VII. Anti-protein VII sera detected a protein of 26 kDa in transfected or BAdV-3-infected cells, which localizes to nucleus and nucleolus of infected/transfected cells. Analysis of mutant protein VII identified four redundant overlapping nuclear/nucleolar localization signals as deletion of all four potential nuclear/nucleolar localization signals localizes protein VII predominantly to the cytoplasm. The nuclear import of protein VII appears to use importin α (α-1), importin-β (β-1) and transportin-3 nuclear transport receptors. In addition, different nuclear transport receptors also require part of protein VII outside nuclear localization sequences for efficient interaction. Proteomic analysis of protein complexes purified from recombinant BAdV-3 expressing protein VII containing Strep Tag II identified potential viral and cellular proteins interacting with protein VII. Here, we confirm that protein VII interacts with IVa2 and protein VIII in BAdV-3-infected cells. Moreover, amino acids 91–101 and 126–137, parts of non-conserved region of protein VII, are required for interaction with IVa2 and protein VIII, respectively.
Full article
(This article belongs to the Section Animal Viruses)
►
Show Figures

Figure 1








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}